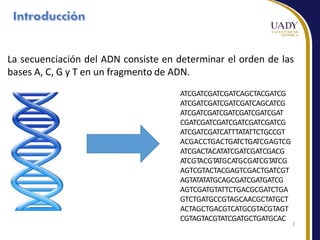
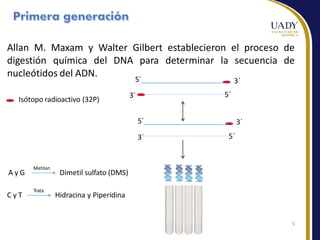
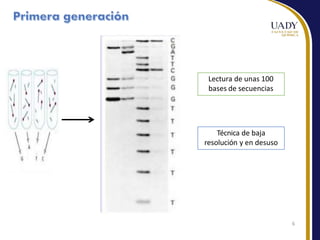
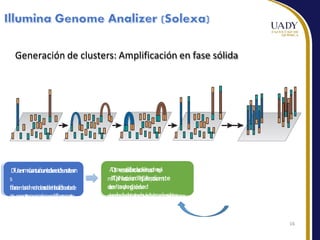
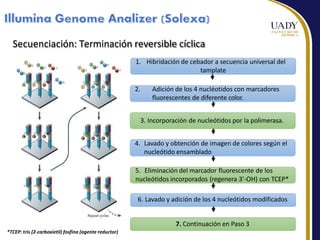
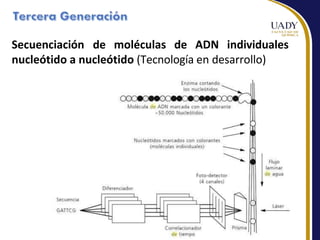
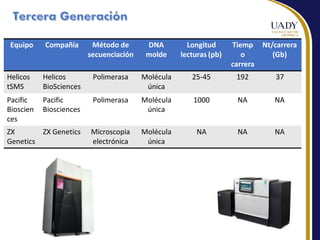
Este documento describe las principales tecnologías de secuenciación masiva de ADN, incluyendo el método de Sanger, secuenciación por terminación reversible cíclica y nanoporos. Explica los pasos de preparación de la muestra, generación de clusters, secuenciación e interpretación de datos para cada método. También compara ventajas y desventajas de las diferentes plataformas de secuenciación.