
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a SERIE BLANCA Y PLAQUETAS.pptx
Similar a SERIE BLANCA Y PLAQUETAS.pptx (20)
Más de DanielZambrano83567
Más de DanielZambrano83567 (20)
Último
Último (20)
SERIE BLANCA Y PLAQUETAS.pptx
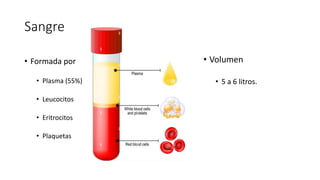
- 1. Sangre • Formada por • Plasma (55%) • Leucocitos • Eritrocitos • Plaquetas • Volumen • 5 a 6 litros.
- 2. PLASMA • Formada por • 90% agua • 10% Proteínas y otros solutos • Albumina • Globulinas • Fibrinógeno
- 6. VALORES NORMALES LEUCOCITOS: 4000-11.000/uL FORMULA LEUCOCITARIA: • Neutrófilos: 2000 – 7000 _____ 60% - 65% • Eosinófilos: 0-400 ______ 0% - 0,4% • Basófilos: menor 1% _______ 0.1% 1% • Linfocitos: 2000- 2500 _______ 20% - 25% • Monocitos: 400 -1000 _______ 0.3% - 0.8%
- 7. PATOLOGIA LEUCOCITOSIS: mayor a 11.000/uL LEUCOPENIA: menor de 4000uL
- 8. NEUTROFILOS Más abundantes Núcleo multilobulado Función: fagocitosis
- 9. EOSINOFILOS Núcleo bilobulado Citoplasma: rojizo (ácido) F: parasitosis, enfermedad crónicas Eosinofilia: mayor de 500uL Eosinopenia: menor de 50uL
- 11. LINFOCITO Tipos: Linfocitos: B- T- NK PATOLOGÍA: Linfocitosis mayor de 5000 Linfopenia: menor 1000
- 12. MONOCITO Núcleo con forma de riñon Es el precursor de los macrófagos. Monocitosis: mayor de 800/uL Monocitopenia: menor de 100uL
- 13. Tema: Pruebas de laboratorio clínico de PLAQUETAS 1. Conteo de plaquetas 2. Valores referenciales normales 3. Cascada de coagulaciones 4. Factores de coagulación 5. Trombosis 6. Actividades
- 14. COAGULOGRAMA • El coagulograma comprende un conjunto de pruebas que exploran la participación de todos los componentes de la hemostasia: • endotelio vascular, • actividad plaquetaria, • plasma y • factores fibrinolíticos.
- 15. COAGULOGRAMA • El coagulograma es un conjunto de pruebas que evalúan de forma global y orientadora el funcionamiento de los diferentes componentes del sistema hemostático. • Su importancia radica profundamente en la sencillez de su realización y en la disponibilidad de los medios y recursos para su ejecución.
- 16. COAGULOGRAMA • Indicaciones: • Detección de trastornos hemorrágicos • Pacientes con clínica sugestiva de CID • Seguimiento de la terapia anticoagulantes y antagonistas de la vitamina-K • Enfermedades hepáticas
- 17. COAGULOGRAMA • Este conjunto de pruebas está integrado por: 1. Tiempo de sangrado 2. Recuento de plaquetas 3. Tiempo de coagulación, 4. Tiempo de protrombina (TP), 5. Tiempo parcial detromboplastina activado (TPTA) y 6. Tiempo de trombina (TT).
- 18. PLAQUETAS Residuos citoplasmáticos de los megacariocitos. Contiene: serotonina, FCP, fibrinógeno Función: hemostasia vida: 10 días. VALOR NORMAL: 150.000 – 400.000 Patología: Trombocitopenia Trombocitosis
- 19. PLAQUETAS Trombocitopenia: • LEVE: 150.000 – 70.000 • MODERDA: 70.000 – 20.000 • SEVERA: MENOR DE 20.000
- 20. COAGULOGRAMA • El tiempo de protombina (TP) es un examen de sangre que mide el tiempo que tarda el plasma de la sangre en coagularse. • Mide la función de la vía de coagulación extrínseca y comunes
- 21. COAGULOGRAMA • Tiempo de sangrado: es el tiempo entre la realización de una pequeña incisión en la piel, el cual produce un sangrado hasta el momento del cese del sangrado. • Es una prueba compleja de realizar: subjetiva. • Que mide: interacción de las plaquetas con la pared de los vasos hasta la formación del coágulo.
- 22. COAGULOGRAMA • TTPa o tiempo parcial activada de tromboplastina, mide el tiempo que tarda en formarse un coágulo de sangre • Mide la función de la vía de coagulación intrínseca y factores de la vía común. • TT o tiempo trombina, mide el paso final de la vía común de la coagulación, la conversión del fibrinógeno en fibrina.
- 23. COAGULOGRAMA • VALORES NORMALES: 1. tiempo de protrombina (TP): 12 a 14 segs. 2. Tiempo de trombina: 18 a 25 segs. 3. tiempo parcial de tromboplastina activado (TPTA): 20 a 40segs con una media de 35segs. 4. INR: mas/menos: 2
- 24. COAGULOGRAMA • INR: Razón internacional normalizada • Valor: mas/menos: 2 • Uso: Valor de referencia para vigilar el uso de anticoagulantes.
- 25. Productos de fragmentación de fibrinógeno • Fibrinólisis: degradación de la fibrina al activarse la enzima: plásmina. Plasminógeno Plasmina Fragmenta: - fibrinógeno - fibrina Patológica: • Primaria: fibrinólisis primaria • Secundaria: CID, Ca, infecciones, ejercicio intenso, etc Laboratorio: -detectables solo en casos patológicos. - tratamiento trombólitico
- 26. Productos de fragmentación de fibrinógeno • DÍMERO- D: -Origina: degradación de la fibrina. -Los valores: son un índice de activación de fibrina en la circulación -Es formado por la acción secuencial de tres enzimas: trombina, Factor XIIIa y plasmina. Valores: 500ng/ml Patologia: -ACV, infecciones, CID, trombolisis fisiológica y terapéutica.
- 27. FACTORES DE COAGULACIÓN • Son todas aquellas PROTEÍNAS que participan y forman parte del coágulo sanguíneo. • Son 13 los factores de coagulación. • Necesitan de cofactores de activación como el calcio, fosfolípidos. I: Fibrinógeno IX: Factor antihemofílico B, Factor de Christmas X : Factor de Stuart-Prower II: Protrombina III: Factor tisular (Tromboplastina) IV: Calcio V: Proacelerina (factor labil) VII: Proconvertina (factor estable) VIII: Factor antihemofílicoA XI: Factor antihemofílico C XII: Factor Hageman XIII: Factor estabilizante fibrina
- 28. III F. de Stuart-Prower F. antihemofílico B II IV Proacelerina F. antihemofílicoA F. antihemofílico C F. Hageman
- 29. ENFERMEDADES ASOCIADAS A FACTORES DE COAGULACIÓN Deficiencia del factor II Deficiencia del factor V Deficiencia del factor VII Deficiencia del factor X Deficiencia del factor XII HemofiliaA Hemofilia B El factor VIII: Su ausencia produce Hemofilia A. El factor IX: Su ausencia provoca Hemofilia B. El factor XI: Su ausencia provoca Hemofilia C.
- 30. Déficit: el coágulo no podrá formarse con facilidad, lo que dará lugar a una hemorragia excesiva. Disminución: Elevación: -hepatopatías graves -Embarazo -CID -Infecciones -Fibrinolisis -Inflamaciónes protrombóticas -IAM FACTOR I FIBRINÓGENO Valores: 200 – 400mg/ml
- 31. FACTOR II: PROTROMBINA Duración de la vida media:60-70hrs Masa (KDa): 72 Nivel en plasma(mg/dl):250-400 Función: se convierte en trombina por la acción del factor Xa Depende de la vitamina K: si sitio de síntesis del factor: hígado Vía: ambas PRUEBAS * Análisis del factor II * Tiempo parcial de tromboplastina * Tiempo de protrombina (TP)
- 32. DEFICIENCIA DE FACTOR II (PROTROMBINA) Es un trastorno muy raro que ocasiona una coagulación deficiente de la sangre. Causada por: * Deficiencia de vitamina K por el uso prolongado de antibióticos, obstrucción de las vías biliares y absorción inadecuada de los nutrientes por parte del tracto intestinal ( malabsorción intestinal) * Enfermedad hepática grave * Uso de anticoagulantes Algunos bebés nacen con deficiencia de vitamina K.
- 33. FACTOR III: TROMBOPLASTINATISULAR Función: se libera con el daño celular Vía: extrínseca Normal: El factor tisular está ausente en las células endoteliales. Sin embargo, cuando se produce la rotura de un vaso sanguíneo, por ejemplo a consecuencia de una herida, el factor tisular de los fibroblastos entra en contacto con la sangre, y además se expresa en las células
- 34. Factor IV : Calcio Masa (KDa): 40 Nivel en plasma(mg/dl): 4-5 Función: median la unión de los factores IX,X ,VII y II. Sitio de síntesis del factor: hígado Vía: ambas
- 35. Factor V : Proacelerina o F. Labil Duración de la vida media: 12 a 16 horas Masa (KDa):350 Nivel en plasma(mg/dl):1 Función: potencia la accion de Xa sobre la protrombina sitio de síntesis del factor: Vía: ambas La deficiencia: hemorragias, Mutaciones: trombosis
- 36. Factor VI: variante activada del Factor V Función: forma activa del factor V Sitio de síntesis del factor: hígado Vía: ambas intermediario
- 37. DEFICIENCIA DEL FACTOR V PRUEBAS • Análisis del factor V que muestra disminución de la actividad • Tiempo de trombina normal • Tiempo parcial de tromboplastina prolongado • Tiempo de protrombina prolongado • Tiempo de sangría ligeramente prolongado
- 38. Duración de la vida media: 8 a 12 horas Nivel en plasma(mg/dl):0.1-0.2 Función: indispensable para la acción del factor X. Su ausencia: hemofilia A Sitio de síntesis del factor : endotelio Función: actúa como uno de los cofactores de la cascada. Laboratorio: TTPa: prolongado TP: normal Tiempo de sangrado: normal la coagulación. FACTOR VIII :F .ANTIHEMOFÍLICO A
- 39. Deficiencia del factor VIII de la coagulación. Causado por un rasgo hereditario recesivo ligado al cromosoma X SÍNTOMAS: Sangrado al interior de las articulaciones y el correspondiente dolor y edema, sangre en la orina o en las heces, hematomas, hemorragias de vías urinarias y digestivas, sangrado prolongado producido por heridas, extracciones dentales y cirugía, sangrado espontáneo. FACTOR VIII :F .ANTIHEMOFÍLICOA
- 40. Deficiencia del factor IX de la coagulación. Enfermedad: Hemofilia - B Causado por un rasgo hereditario recesivo ligado al cromosoma X Sintomatología similar a la Hemofilia A. Tratamiento: Similar a Hemofilia A, sólo que infundiendo factor IX. Duración de la vida media: 18 -24 horas Nivel en plasma(mg/dl):0.3 Función:Activado por el factor XIa en presencia de Ca. Depende de la vitamina K:si sitio de síntesis del factor : hígado Vía: intrínseca FACTOR IX
- 41. Factor X: Factor Stuart -autoprotrombina III Duración de la vida media: 1 a 2 días Nivel en plasma(mg/dl): 1 Función: activado por el complejo IXa-VIII-Ca en la via intrínseca o por VII- III-ca en la extrínseca Depende de la vitamina K: si Sitio de síntesis del factor: higado Vía: ambas
- 42. • Es una serina proteasa • Duración de la vida media 52 horas • Nivel en plasma(mg/dl): 0.5 • Función: convertido en la proteasa XIa por la acción del factor XIIa; XIa activa al factor IX • Sitio de síntesis del factor: higado Vía: intrinseca Factor de coagulación XI o antecedente tromboplastínico del plasma
- 43. TROMBOSIS Adulto mayor: factores de riesgo TVP Joven: genéticas, hereditarias. Trombofilia: tendencia a la trombosis por defecto en el equilibrio coagulación- anticoagulación.
- 44. • Exámenes: • Proteina C • Proteína S • Antitrombina III • Mutación del Factor V • Homocisteína en sangre • Anticoagulante Lúpico Trombofilia: tendencia a la trombosis por defecto en el equilibrio coagulación- anticoagulación.
- 45. • Proteína C • Hereditario • Observa: neonatos con púrpura fulminante • Causas: Insuficiencia hepática, CID. • Proteína S • Cofactor de la proteína C • Déficit: hereditario y adquiridas: varicela, embarazo, Sd. Nefrótico, enfermedad hepática, LES, uso de ACO. • Anticoagulante Lúpico: • Son auto-anticuerpos que se une a las proteínas que contienen fosfolípidos. • Patologías: LES, Sd. Antifosfolípidico • Sospecha: trombocitopenias • Homocisteína en sangre • Aminoácido. • Sintetizado: metionina, requiere de vitamina B6 y B12 • Valores: 5 -16umol/L • Patologia: déficit de vitaminas, genéticas • Mecaismo: alteración endotelial asociado a: ACV, EAP, TVP, cardiopatías isquémicas.
Notas del editor
- 1uL: millonésima parte de un 1 litro. También llamado microlitro
- 1uL: millonésima parte de un 1 litro. También llamado microlitro
- Son una parte vital para la investigación de defectos en la hemostasia, seguimiento de tratamientos con trastornos trombóticos o hemorrágicos.
- Son una parte vital para la investigación de defectos en la hemostasia, seguimiento de tratamientos con trastornos trombóticos o hemorrágicos.
- TPT, tiempo parcial de tromboplastina, mide el tiempo que tarda en formarse un coágulo de sangre El tiempo de protombina (TP) es un examen de sangre que mide el tiempo que tarda la porción líquida de la sangre (plasma) en coagularse.
- Forma: discoidal, biconvexo Tamaño: 2-3mc
- Forma: discoidal, biconvexo Tamaño: 2-3mc
- TPT, tiempo parcial de tromboplastina, mide el tiempo que tarda en formarse un coágulo de sangre El tiempo de protombina (TP) es un examen de sangre que mide el tiempo que tarda la porción líquida de la sangre (plasma) en coagularse.
- El TS mide la fase vascular y plaquetaria de las etapas tempranas de la hemostasia.
- TPT, tiempo parcial de tromboplastina, mide el tiempo que tarda en formarse un coágulo de sangre El tiempo de protombina (TP) es un examen de sangre que mide el tiempo que tarda la porción líquida de la sangre (plasma) en coagularse.
- La trombina es un enzima de la coagulación que convierte el fibrinógeno en fibrina, la cual es degradada por otro sistema de defensa, denominado sistema fibrinolítico, con formación de un producto de degradación que se conoce como dímero D
- Fase inicial El complejo factor tisular-factor VII, de forma directa e indirectamente a través del factor IX, activa inicialmente el factor X transformando pequeñas cantidades de protrombina en trombina, que son aún insuficientes para completar el proceso de formación de la fibrina. Fase de amplificación La trombina así formada, junto con el calcio de la sangre y los fosfolípidos ácidos, que provienen de la plaqueta, participa activamente en un proceso de retroalimentación para la activación de los factores XI, IX, VIII y V y, de forma especial aceleran la activación de la plaqueta. Simultáneamente, por mecanismos quimiotácticos, los factores mencionados son atraídos a la superficie de las plaquetas donde tienen lugar de forma muy rápida importantes procesos de activación y multiplicación. Fase de propagación La amplificación del proceso por mecanismos de retroalimentación entre trombina y plaqueta y la activación de todos estos factores permiten activar grandes cantidades del factor X y formar el complejo protrombinasa para convertir la protrombina en trombina y, a expensas de ésta, el fibrinógeno en fibrina. El proceso final, siempre en la superficie de la plaqueta, se acelera para generar de forma explosiva grandes cantidades de trombina y fibrina. La nueva cascada de la coagulación presenta la formación de fibrina como resultado conjunto de dos procesos: coagulación (representado por la trombina) y actividad de la plaqueta, que mutuamente se complementan. La inhibición profunda y combinada de ambos procesos conduce necesariamente a hemorragias severas,
- Tiempo de sangrado: prolongado
- Es una glicoproteína de membrana ,presente en los fibroblastos de la pared de los vasos sanguíneos y en otras células (por ejemplo, los monocitos).
- El factor V Leiden es el trastorno de hipercoagulabilidad hereditario
- Proteína c: se activa por la trombina, es dependiente de vitamina k