Descargado 147 veces



















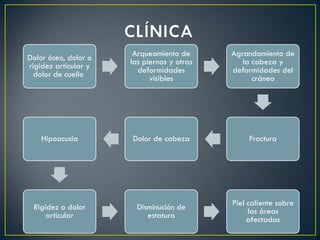

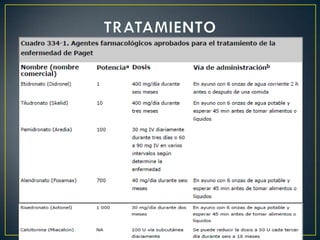








La osteogénesis imperfecta es una enfermedad congénita caracterizada por huesos frágiles que se rompen fácilmente, incluso sin causa aparente. Se debe a un fallo genético que causa una insuficiente formación del colágeno en el cuerpo. Puede presentarse en varios tipos que difieren en gravedad y manifestaciones clínicas. No tiene cura, pero el tratamiento incluye fisioterapia, cirugía y medicamentos para fortalecer los huesos.






















































































