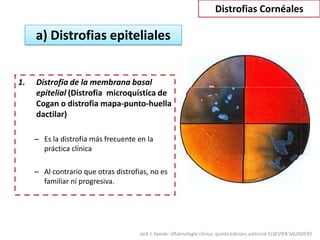
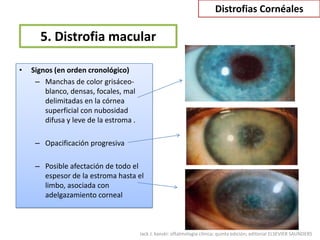
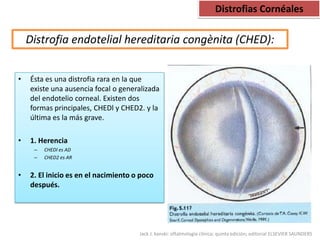
Las distrofias cornéales son alteraciones progresivas de la transparencia corneal que se presentan sin inflamación. Se clasifican en epiteliales, de la capa de Bowman, del estroma y endoteliales. La distrofia más frecuente es la distrofia de la membrana basal epitelial, la cual causa erosiones corneales recurrentes desde la segunda década de vida.