


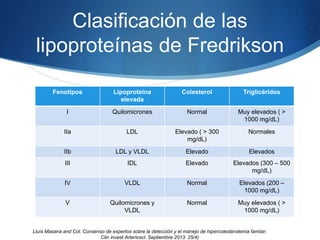





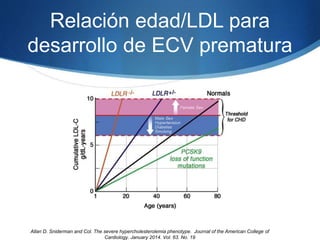
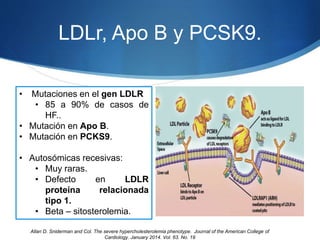

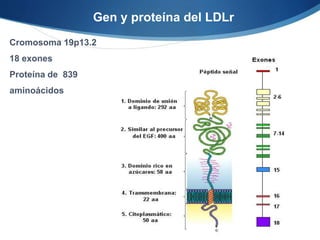


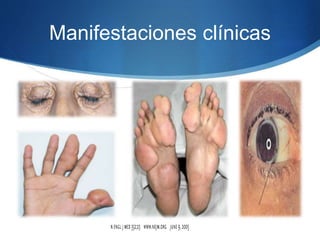










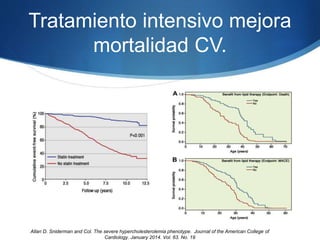


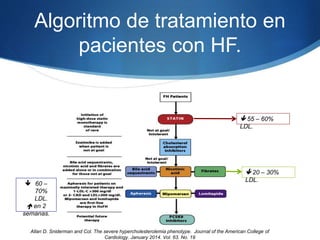

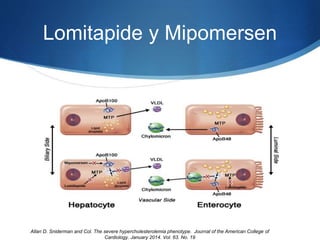
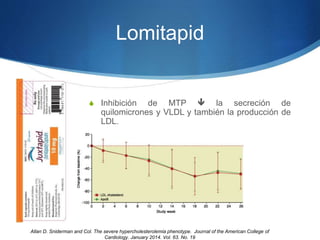



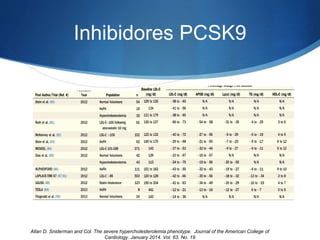


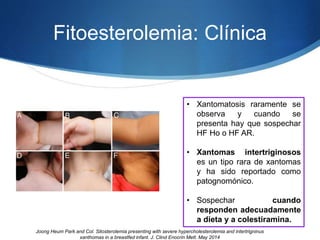
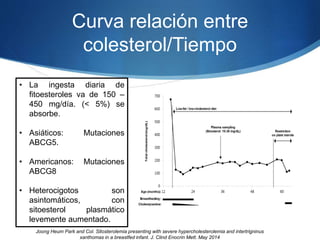




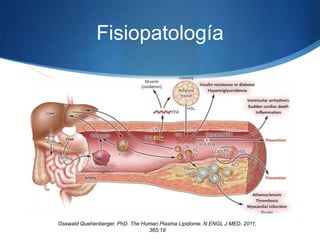
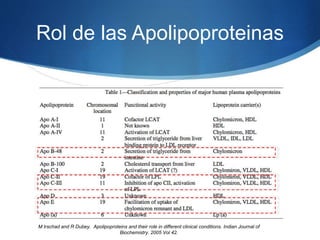


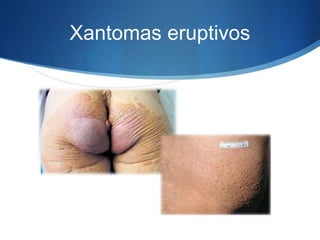

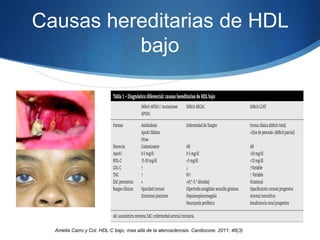


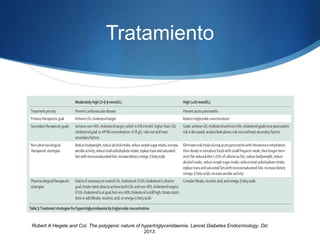






El documento aborda las dislipidemias primarias, haciendo énfasis en la hipercolesterolemia familiar, un trastorno genético que se presenta con concentraciones elevadas de colesterol LDL y un riesgo alto de enfermedad coronaria. Se describen las clasificaciones de hiperlipidemias, los criterios diagnósticos y recomendaciones de tratamiento que incluyen cambios en el estilo de vida y el uso de medicamentos. Se destacan terapias recientes como lomitapide y mipomersen, que han demostrado eficacia en la reducción de LDL en pacientes con hipercolesterolemia familiar.



















































































