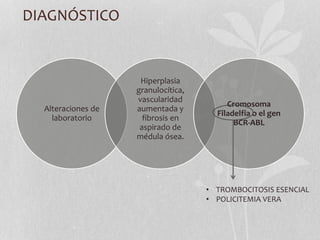
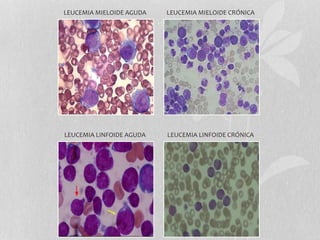
Este documento presenta información sobre las leucemias. Discute la epidemiología de las leucemias en los Estados Unidos para 2014, con alrededor de 52,380 nuevos casos y 24,090 muertes. Describe las clasificaciones de las leucemias agudas y crónicas, y proporciona detalles sobre la leucemia mieloide aguda y crónica, incluidos factores de riesgo, cuadros clínicos, pronóstico y tratamiento.



















































































![C:\fakepath\leucemia[1]](https://cdn.slidesharecdn.com/ss_thumbnails/cfakepathleucemia1-100819231029-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)





































