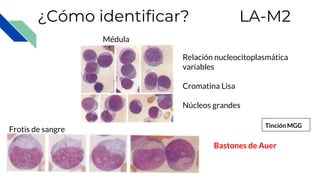
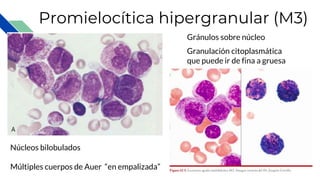
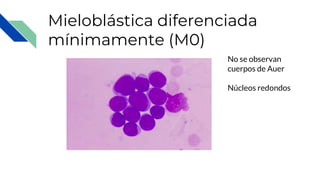
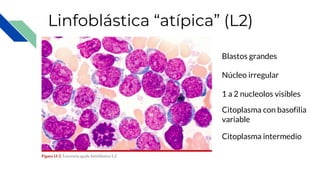
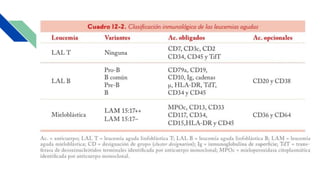
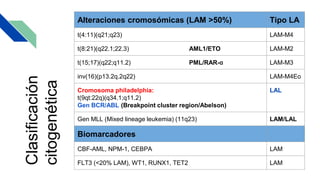
Este documento proporciona información general sobre la leucemia aguda, incluyendo su definición, causas, síntomas, diagnóstico y tratamiento. La leucemia aguda es un cáncer de la sangre que causa la proliferación desordenada de células blancas de la sangre. Se diagnostica mediante exámenes de sangre y médula ósea y su tratamiento involucra quimioterapia, con el objetivo de inducir la remisión y prevenir recaídas a través de tratamientos de mantenimiento a largo