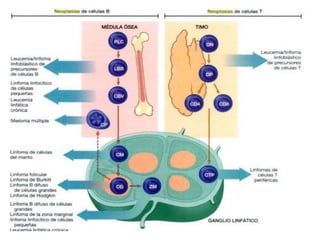
El documento describe los linfomas, un grupo heterogéneo de neoplasias malignas del sistema linfático. Existen dos principales tipos de linfoma: el linfoma de Hodgkin y el linfoma no Hodgkin. El linfoma de Hodgkin se caracteriza por la proliferación tumoral de los ganglios linfáticos, mientras que el linfoma no Hodgkin comprende una variedad de tumores que afectan las células del sistema inmune. El documento proporciona detalles sobre la epidemiología, etiología, clasificación, diagn
















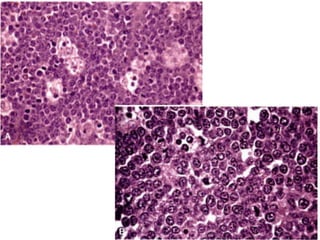
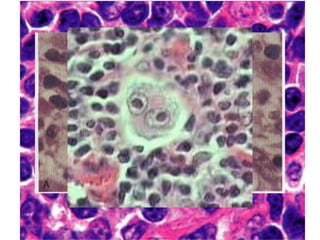
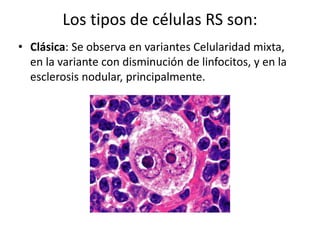
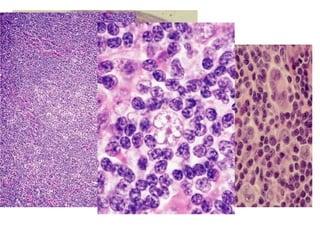
![• El componente reactivo (linfocitos pequeños maduros
[B: alrededor de folículos, T: alrededor de ceulas RS],
eosinófilos, plasmocitos, histiocitos, células foliculares
dendríticas, neutrófilos, fibroblastos, fibras colagenas,
fibrosis y capilares).](https://image.slidesharecdn.com/linfomas-170508012827/85/Linfomas-21-320.jpg)







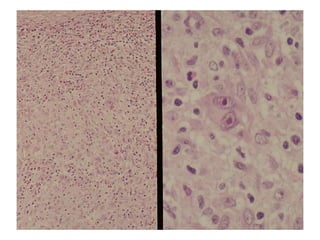

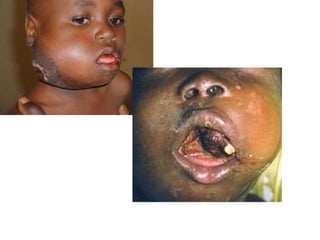
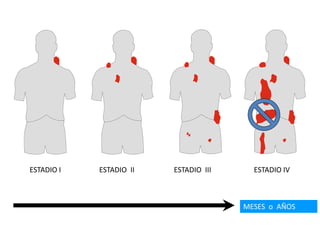
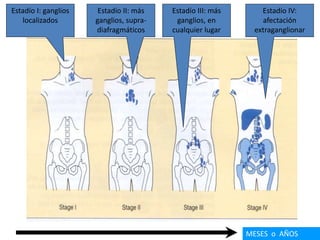
![• Linfadenopatía asintomática (80%), Este agrandamiento de
los ganglios linfático puede causar secundariamente
síntomas por compresión de vena, nervio, o el estomago.
La ictericia intrahepática, Disnea, tos y sibilancias por
compresión traqueobronquial, también la esplenomegalia y
hepatomegalia. Dolor por alcohol.
• " B "( pérdida de peso inexplicable [> 10% del peso corporal
en los últimos 6 meses] y apetito, en un 10% del peso total ,
fiebre, sudores nocturnos
• fiebre de Pel-Epstein
• sindromes paraneoplasicos, incluyendo la degeneración
cerebelosa, neuropatía, síndrome de Guillain-Barré, o
leucoencefalopatía multifocal.](https://image.slidesharecdn.com/linfomas-170508012827/85/Linfomas-40-320.jpg)


















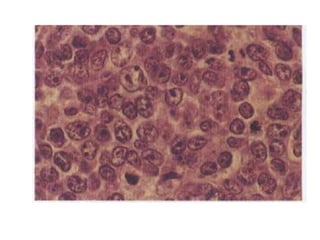





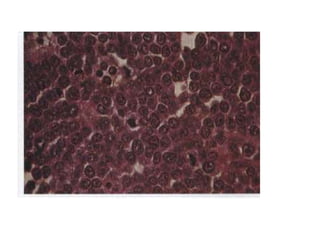

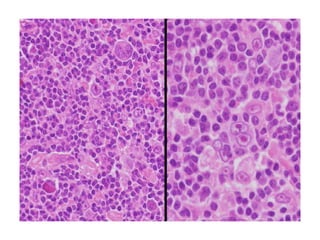
![INMUNOFENOTIPO
• Tumores de células B relativamente maduras que
expresan en su superficie IgM, cadenas ligeras
monotipicas κ y λ. CD19,20,10 +
GENETICA
• Todos se caracterizan por traslocaciones del gen
c-myc situado en el cromosoma 8. Su pareja suele
ser el locus de las IgH [t(8;14)] pero también
puede ser el locus de las cadeas ligeras κ [t(2;8)]
y λ [t(8;22)].](https://image.slidesharecdn.com/linfomas-170508012827/85/Linfomas-71-320.jpg)