Descargado 52 veces












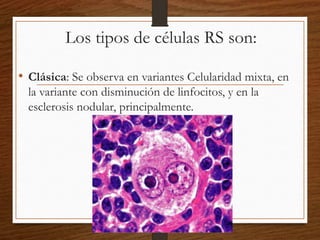















![• Pérdida de peso inexplicable [> 10% del peso corporal en los
últimos 6 meses] y apetito, en un 10% del peso total , fiebre, sudores
nocturnos
• Fiebre de Pel-Epstein
• Síndromes paraneoplasicos, incluyendo la degeneración cerebelosa,
neuropatía, síndrome de Guillain-Barré, o leucoencefalopatía
multifocal.](https://image.slidesharecdn.com/linfomahodkin-140710163326-phpapp01/85/Linfoma-Hodkin-29-320.jpg)















El documento describe los linfomas, incluyendo las diferencias entre el linfoma de Hodgkin y los linfomas no Hodgkin. Explica que los linfomas son neoplasias malignas que se originan en el sistema linfoide y pueden ser nodales u extranodales. También describe las clasificaciones histológicas, los síntomas, el diagnóstico, el tratamiento y el pronóstico de los diferentes tipos de linfoma.











































































































