Descargado 371 veces
![Tema 2: Estructura y Función de las Proteínas (5/5) Prof. Vanessa Miguel [email_address] @bioquitips Noviembre 2011 Universidad Central de Venezuela Facultad de Medicina Escuela de Medicina Luis Razetti Cátedra de Bioquímica](https://image.slidesharecdn.com/clase5tema22011-vm-111125072731-phpapp02/85/Separacion-y-estudio-de-las-Proteinas-1-320.jpg)

















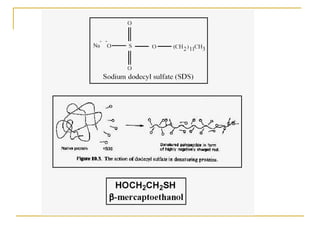






![Salting in (salado) Solubilidad a baja fuerza ionica (0.001 – 0.02 M) aumenta con la [ sales] Salting in Al aumentar la [sales] proteína se rodea de contraiones más soluble por > interacción proteína-solvente](https://image.slidesharecdn.com/clase5tema22011-vm-111125072731-phpapp02/85/Separacion-y-estudio-de-las-Proteinas-54-320.jpg)
![Salting out (precipitación por salado) La solubilidad a alta fuerza iónica ( mayor a 0,02M) disminuye con la [sales] Salting out La alta [sal] remueve la esfera de solvatación de la proteína y éstas precipitan](https://image.slidesharecdn.com/clase5tema22011-vm-111125072731-phpapp02/85/Separacion-y-estudio-de-las-Proteinas-55-320.jpg)


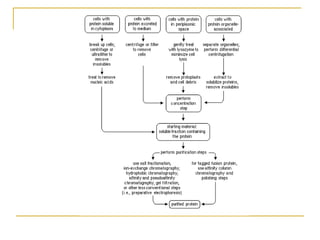
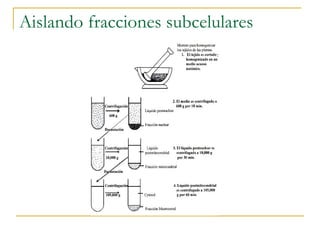
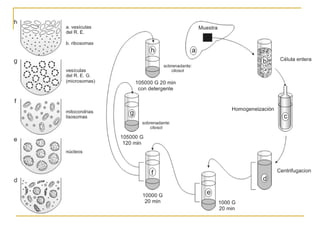
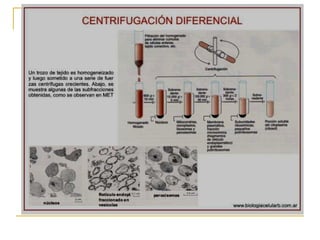
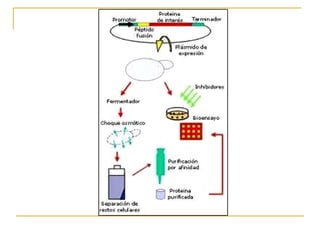
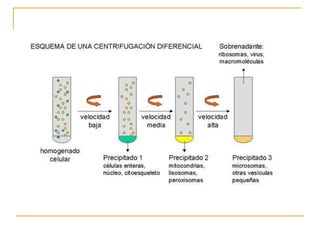
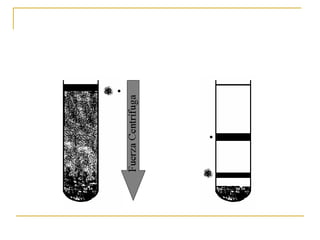
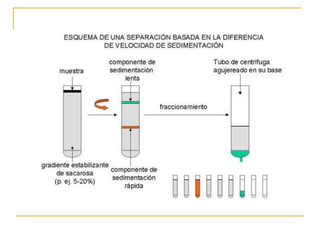
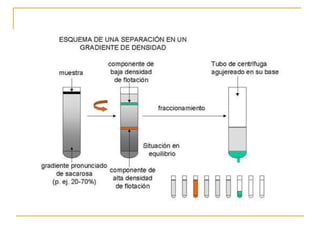
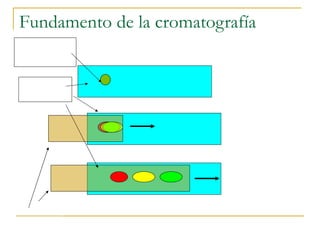
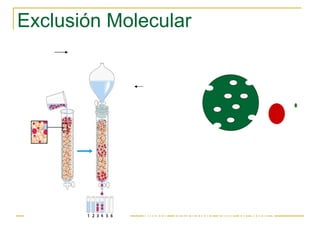
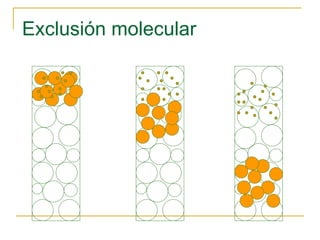
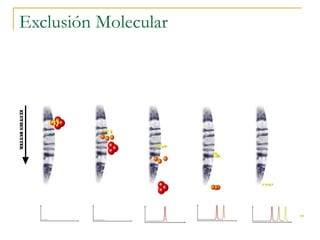
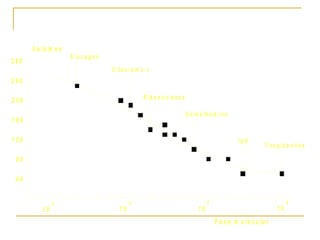
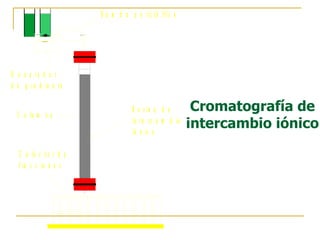
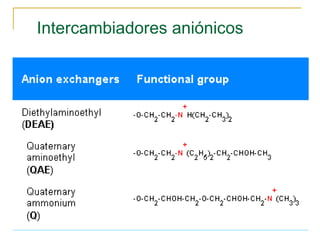
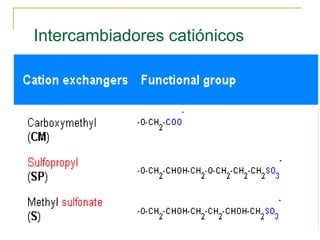
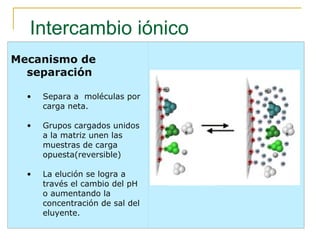
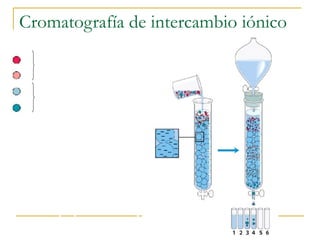
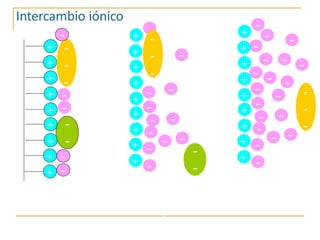
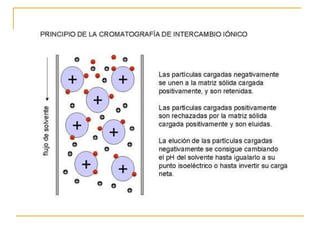
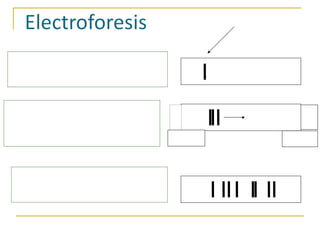
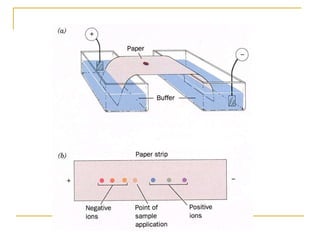
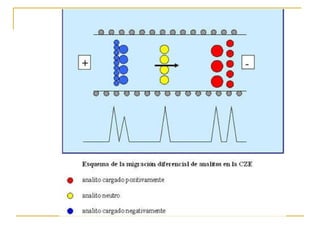
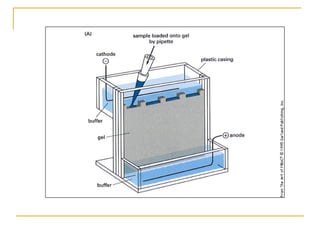
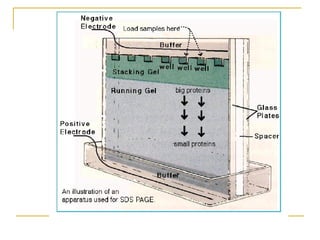
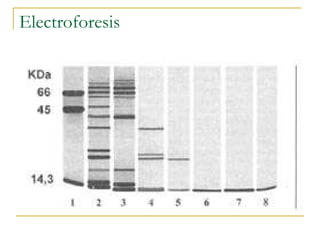
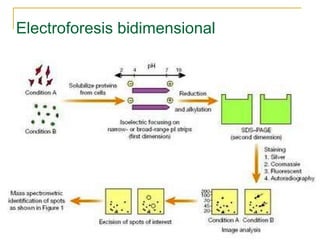
Este documento describe varios métodos para el aislamiento y purificación de proteínas, incluyendo la centrifugación, cromatografía por exclusión de tamaño, intercambio iónico y electroforesis. Explica cómo estas técnicas separan las proteínas basadas en sus propiedades físicas como tamaño, carga eléctrica y solubilidad.




















































![Presentacion evaluaciónaletheiacies2013 [modo de compatibilidad]](https://cdn.slidesharecdn.com/ss_thumbnails/presentacionevaluacinaletheiacies2013mododecompatibilidad-130511082442-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)














