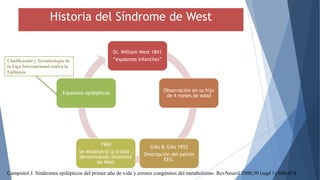
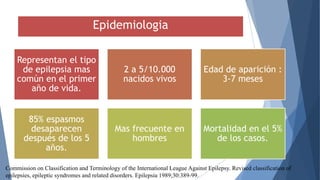
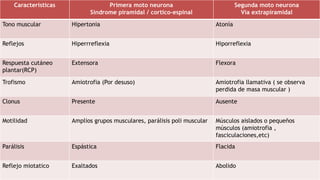
El síndrome de West es una encefalopatía epiléptica caracterizada por espasmos epilépticos, retraso del desarrollo psicomotor y hipsarritmia en el EEG. Su etiología puede ser sintomática, criptogénica o idiopática, representando la forma más común de epilepsia en el primer año de vida, con una incidencia de 2 a 5 por cada 10,000 nacidos vivos. Aunque el pronóstico es generalmente desfavorable, es mejor en casos idiopáticos, donde los niños pueden experimentar una desaparición de crisis y un desarrollo psicomotor normal.