Descargado 857 veces




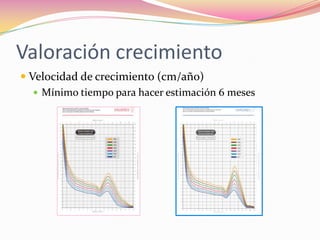
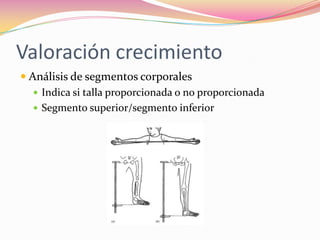
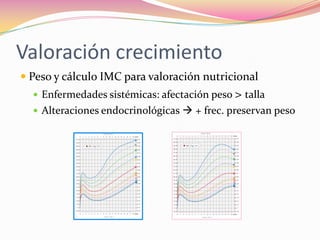


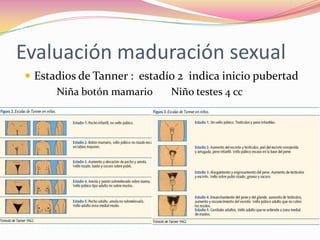
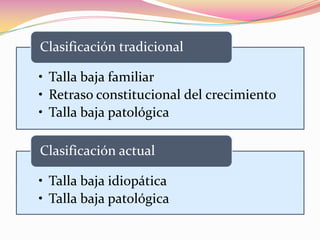
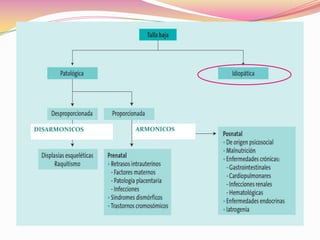




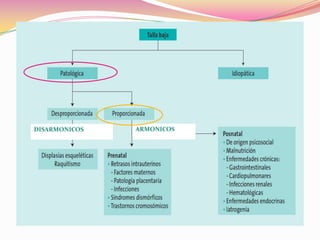



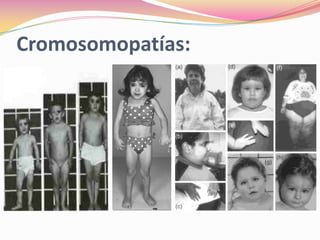





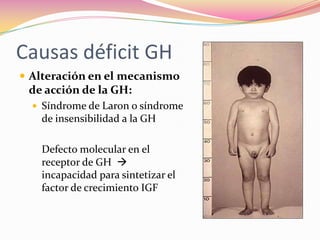



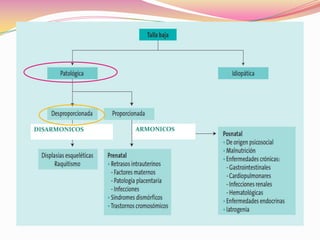

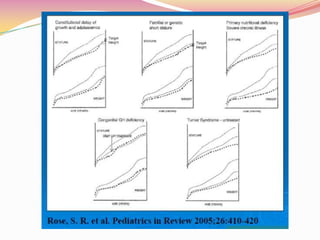








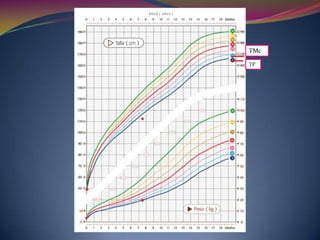
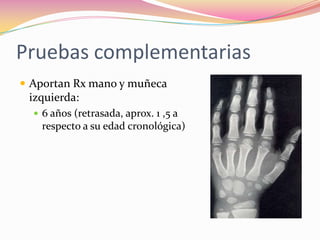
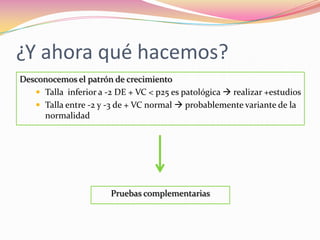
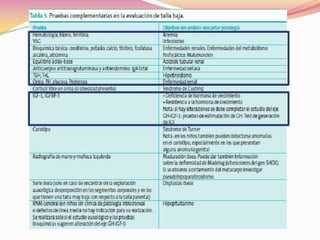





Este documento proporciona información sobre la evaluación del crecimiento en niños. Describe las herramientas para la valoración del crecimiento, incluyendo la longitud, talla, peso, índice de masa corporal, velocidad de crecimiento y maduración ósea. También explica las causas de talla baja, como la idiopática, familiar, déficits hormonales, enfermedades crónicas y síndromes. El objetivo es distinguir niños con retraso patológico del crecimiento de aquellos dentro de la variación normal


































































































