Descargado 20 veces













![DISFUNCIÓN HIPOFISARIA ANTERIOR
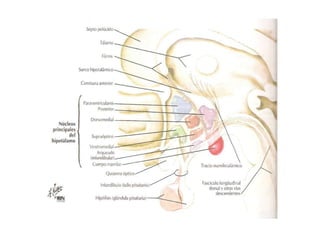
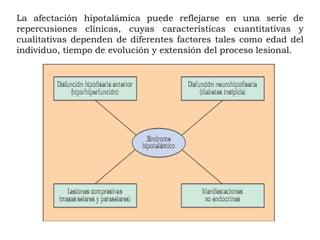
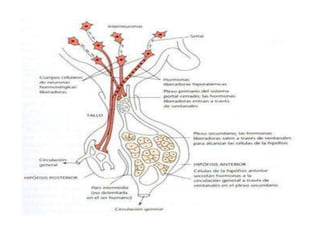
El deficit de secreción de hormonas hipotalámicas
hipofisotropas estimuladoras conduce a una
insuficiencia adenohipofisaria parcial o total, con defecto
de producción de hormona estimuladora del tiroides
[TSH], hormona adrenocorticotropa [ACTH], hormona del
crecimiento [GH] u hormonas gonadotropas[LH, FSH].
Por el contrario, el déficit de secreción de PIH va a
conducir al desarrollo de hiperprolactinemia.
Otras alteraciones más sutiles pueden radicar en una
mera distorsión de los ritmos circadianos de secreciones
hormonales hipotalámicas o en un incremento de la
producción ocasional de hormonas hipofisiotropas
hipotalámicas: pubertad precoz (GnRH), acromegalia
(GHRH), síndrome de Cushing (CRH).](https://image.slidesharecdn.com/02dr-cabrera-sndromedehiperfuncinhipofisiaria-121015015858-phpapp02/85/02-dr-cabrera-sa-ndrome-de-hiperfuncia-n-hipofisiaria-18-320.jpg)



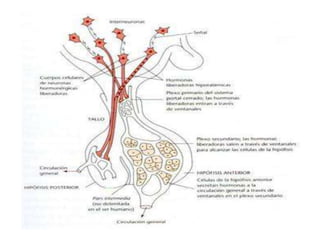


























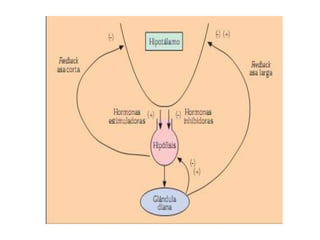
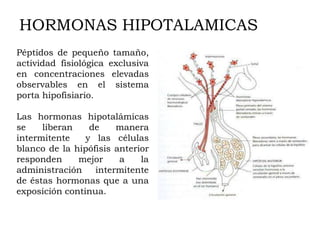
Este documento describe los síndromes de hiperfunción e hipofunción hipotalámico-hipofisarios. Explica cómo los neurotransmisores regulan la síntesis y secreción de hormonas hipotalámicas que a su vez controlan la producción de hormonas hipofisarias. También describe las diversas hormonas hipotalámicas y sus funciones, y los síndromes que pueden resultar de la disfunción hipotalámica o hipofisaria.











































































































