Descargado 655 veces





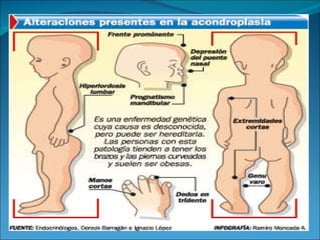


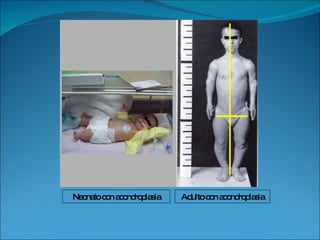



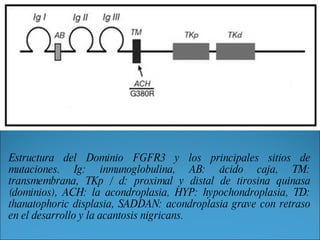



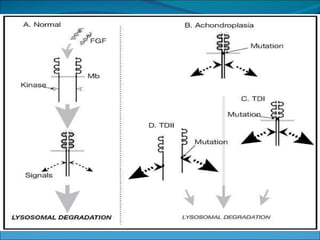




La acondroplasia es una forma de enanismo autosómico dominante que ocurre en 1 de cada 30,000 personas, causada por mutaciones en el gen FGFR3 que codifica el receptor de factor de crecimiento fibroblástico 3. Los acondroplásicos tienen huesos largos no desarrollados adecuadamente y alcanzan una altura predecible de entre 122-144 cm para varones y 117-137 cm para mujeres. No existe tratamiento para aumentar la estatura, solo cirugía de alargamiento de huesos.









































































































