



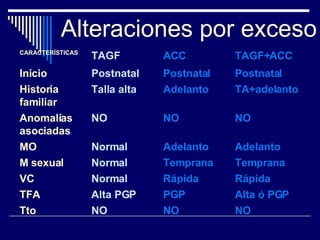






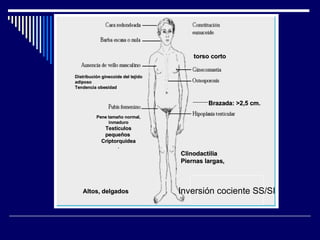
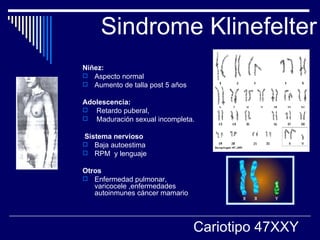
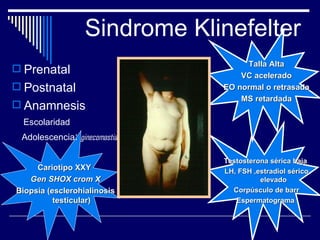




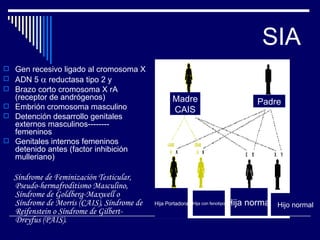


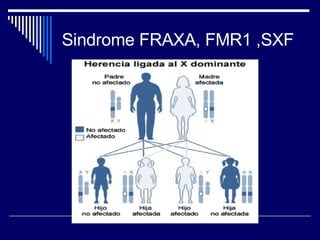



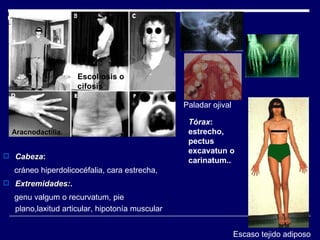




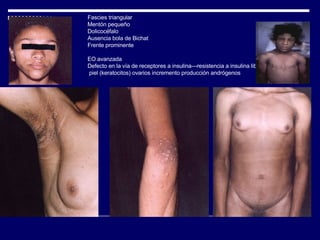

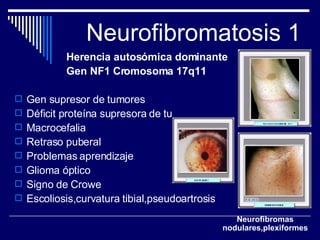
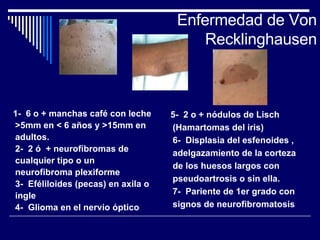



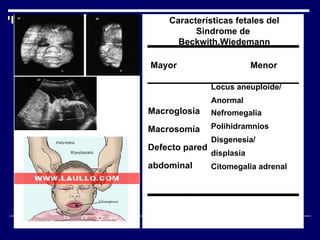






1. El documento describe varias alteraciones que pueden causar un aumento anormal de la talla, incluyendo trastornos genéticos como el síndrome de Marfan y Klinefelter, y trastornos hormonales como la acromegalia. 2. También describe síndromes de hipercrecimiento prenatal como el síndrome de Beckwith-Wiedemann y Soto, que se caracterizan por un crecimiento excesivo en los primeros años de vida. 3. Las alteraciones se clasifican como primarias o secundarias dependiendo de si





















































































