Descargado 24 veces






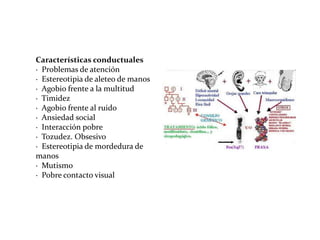




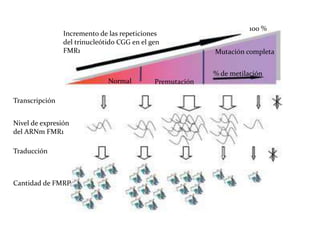



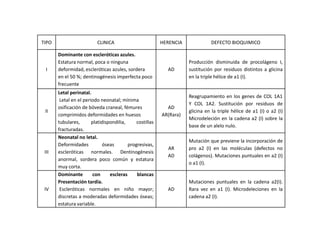

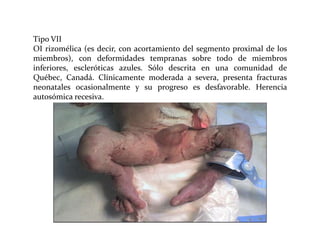






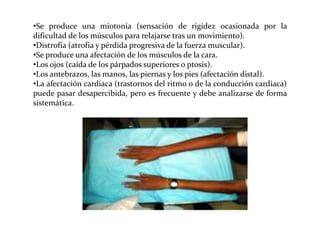
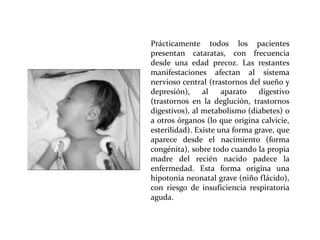

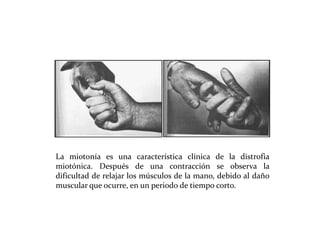

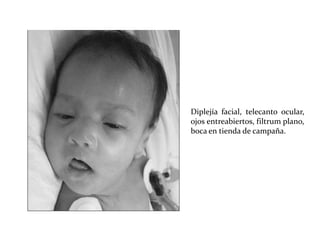

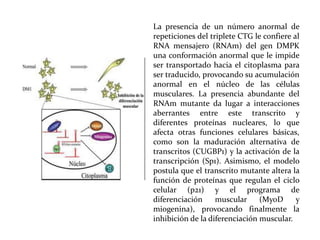
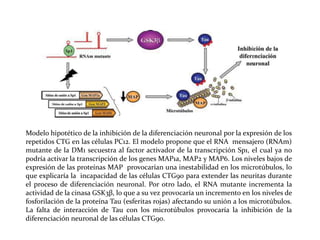
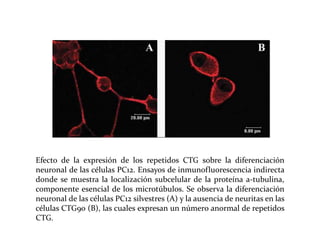


Este documento describe el Síndrome de X frágil, la causa genética más común de retraso mental hereditario. Se debe a una mutación en el gen FMR1 en el cromosoma X que provoca la falta de una proteína crucial. Puede causar desde un retraso leve hasta uno severo asociado a autismo. Afecta más a los hombres que a las mujeres. No tiene cura, pero el diagnóstico y tratamiento temprano pueden mejorar los síntomas.











































































































