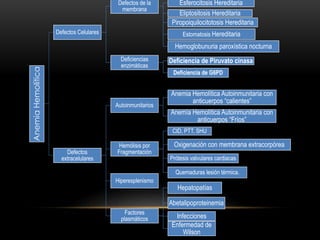
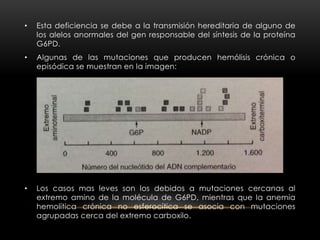
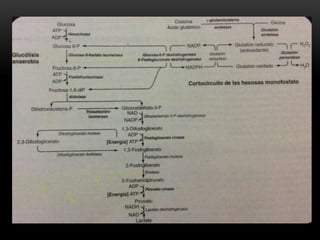
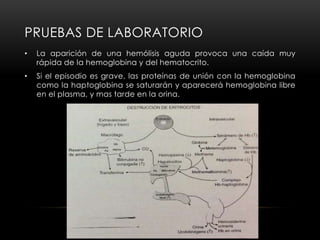
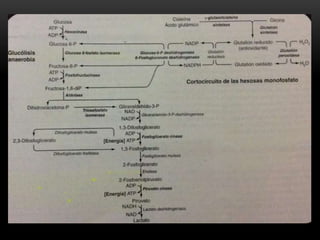
Este documento resume las características de la anemia hemolítica causada por deficiencias de la enzima glucosa-6-fosfato deshidrogenasa (G6PD) y de la piruvato cinasa. Describe cómo estas deficiencias enzimáticas conducen a la destrucción prematura de los eritrocitos, causando anemia hemolítica episódica o crónica. También explica los síntomas, hallazgos de laboratorio y tratamiento de estas deficiencias enzimáticas.