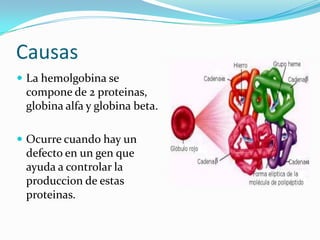
La talasemia es un trastorno hereditario de la sangre que causa anemia debido a un defecto en los genes que controlan la producción de las cadenas alfa y beta de la hemoglobina. Esto provoca la destrucción excesiva de los glóbulos rojos. Existen diferentes tipos dependiendo de qué cadena se ve afectada y la gravedad del defecto genético. El diagnóstico incluye análisis de sangre y estudios genéticos, y el tratamiento puede incluir transfusiones de sangre, suplementos o trasplante de mé