Descargado 1659 veces
























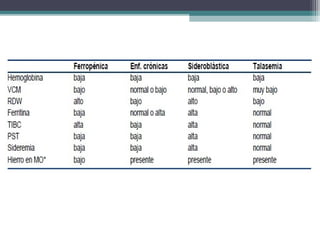
![SIDEROBLÁSTICASIDEROBLÁSTICA
• Uso inadecuado del hierro para formar Hb, a pesar
de tener [] normales de hierro.
• Se caracterizan por la presencia en la MO de
sideroblastos en anillo, aumento del Fe+
circulante
y de depósito y eritropoyesis ineficaz.](https://image.slidesharecdn.com/sindromeanemico2013-130911125729-phpapp02/85/Sindrome-anemico-2013-25-320.jpg)




























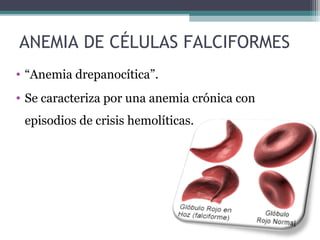





































Este documento describe el síndrome anémico y diferentes tipos de anemia. Define la anemia como una reducción de la masa eritrocitaria o de la concentración de hemoglobina por debajo de los valores normales. Describe las causas de la anemia como pérdida de sangre, producción inadecuada de eritrocitos, y destrucción excesiva. Además, clasifica las anemias y explica las características, diagnóstico y tratamiento de diferentes tipos como la anemia ferropénica, las anemias hemolíticas y las




































![ANEMIA GENERALIDADES.pptx [Reparado] (1).pptx](https://cdn.slidesharecdn.com/ss_thumbnails/anemiageneralidades-250927041105-ef7a9965-thumbnail.jpg?width=640&height=640&fit=bounds)

























































