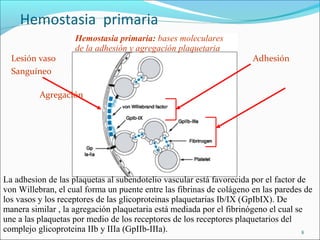
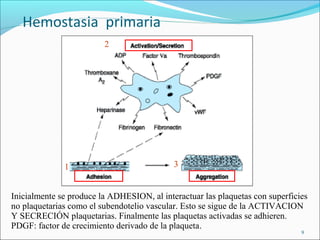
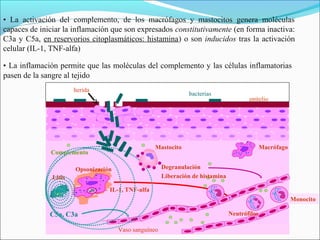
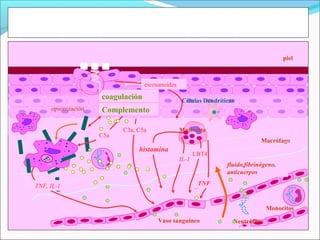
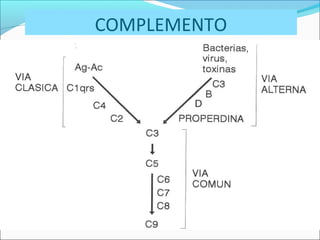
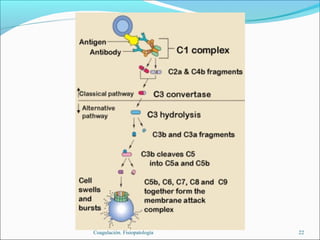
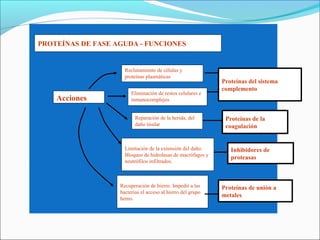
Este documento describe los mecanismos de la coagulación sanguínea, incluyendo la cascada de coagulación, el papel de los vasos sanguíneos, plaquetas y factores de coagulación. También explica los procesos de hemostasia primaria y secundaria, asi como la fibrinólisis. Por último, detalla los componentes y funciones del sistema del complemento.