Descargado 50 veces









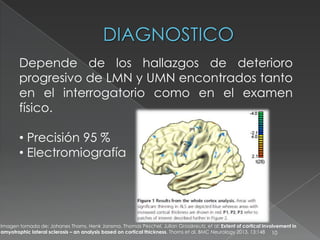











La esclerosis lateral amiotrófica (ELA) es una enfermedad degenerativa de las neuronas motoras, caracterizada por la pérdida progresiva de la función muscular sin dolor ni pérdida de sensación. Aunque su etiología sigue siendo desconocida y no hay cura, la enfermedad afecta principalmente a hombres y su incidencia aumenta con la edad. La expectativa de vida suele ser de unos tres años, siendo la muerte comúnmente causada por complicaciones respiratorias.




































![Leidy[1]](https://cdn.slidesharecdn.com/ss_thumbnails/leidy1-100830103500-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)
























































