Descargado 1843 veces


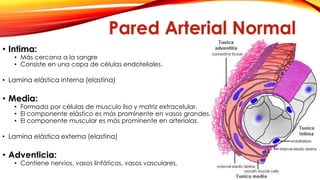
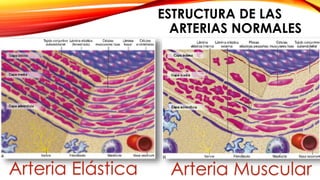



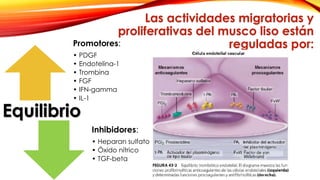
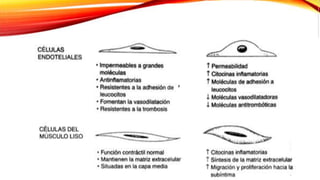

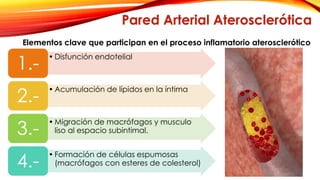


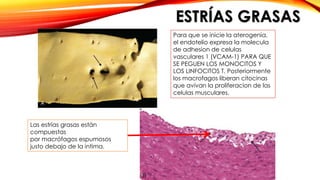
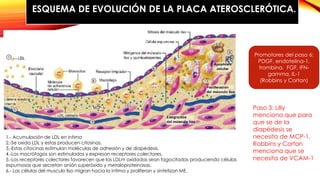

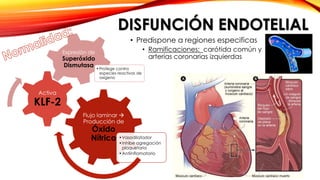
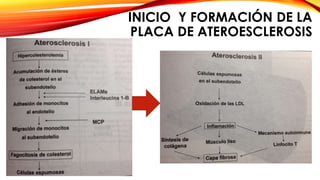

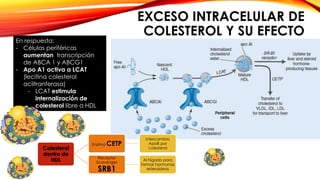
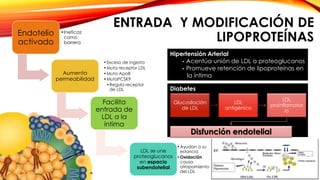

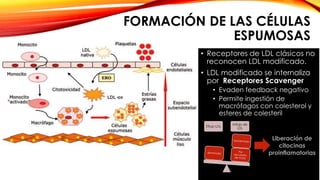
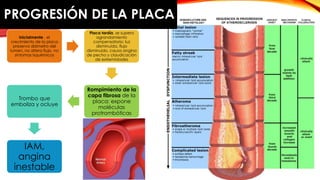

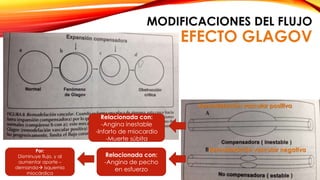
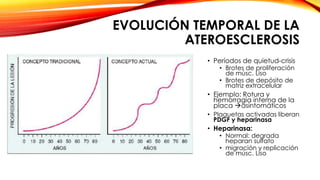
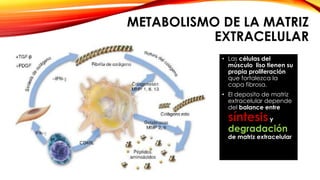
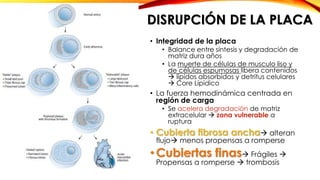

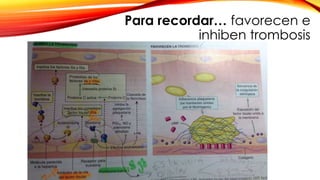
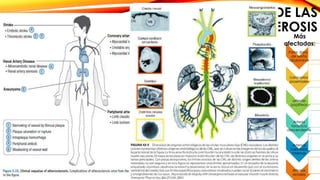




La cardiopatía isquémica se debe a la formación de placas de aterosclerosis en las arterias que disminuyen el flujo sanguíneo al corazón. Esta se inicia por la acumulación y oxidación de colesterol LDL en la pared arterial, lo que causa una inflamación crónica y reclutamiento de macrófagos. Estos forman células espumosas que atraen más colesterol y migrar células musculares lisas, engrosando la placa y obstruyendo el vaso.






























![Arterioesclerosis [Autoguardado].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/arterioesclerosisautoguardado-221105234625-f450b407-thumbnail.jpg?width=640&height=640&fit=bounds)




















































