Fisiopatología gastrointestinal ii
•Descargar como DOCX, PDF•
1 recomendación•773 vistas

cirrosis hepatica

Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Destacado
Similar a Fisiopatología gastrointestinal ii
Similar a Fisiopatología gastrointestinal ii (20)
Último
Último (20)
Fisiopatología gastrointestinal ii
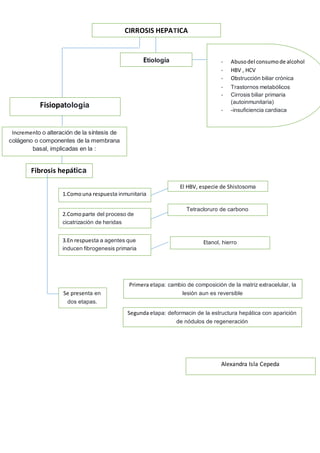
- 1. CIRROSIS HEPATICA Etiología - Abusodel consumode alcohol - HBV , HCV - Obstrucción biliar crónica - Trastornos metabólicos - Cirrosis biliar primaria (autoinmunitaria) - -insuficiencia cardiaca Fisiopatologia Incremento o alteración de la síntesis de colágeno o componentes de la membrana basal, implicadas en la : Fibrosis hepática 1.Comouna respuesta inmunitaria 2.Comoparte del proceso de cicatrización de heridas 3.En respuesta a agentes que inducen fibrogenesis primaria El HBV, especie de Shistosoma Tetracloruro de carbono Etanol, hierro Se presenta en dos etapas. Primera etapa: cambio de composición de la matriz extracelular, la lesión aun es reversible Segunda etapa: deformacin de la estructura hepática con aparición de nódulos de regeneración Alexandra Isla Cepeda
- 2. 1. Debidas a hipertensión portal con derivación portosistemica Ascitis con riesgo aumentado de peritonitis bacteriana espontanea Incremento del riesgo de sepsis Riesgo aumentado de coagulación intravascular diseminada Esplenomegalia con trombocitopenia Encefalopatía Varices Sensibilidad a fármacos Deficiencia de ácidos biliares con malabsorción de grasa, vitaminas liposolubles Hiperglucemia 2. Debidas a perdida de hepatocitos Hipoglucemia Coagulapatia debida a síntesis deficiente de factores de la coagulación Edema periférico debido a hipoalbuminemia Coma hepático Alexandra Isla Cepeda MANIFESTACIONES CLINICAS
- 3. CIRROSIS HEPATICA La cirrosis hepática, es una de las enfermedades más importantes y que más aquejan a este órgano. La cirrosis Hepática es una enfermedad crónica progresiva que consiste en la muerte del tejido hepático normal, que es sustituido por un tejido fibroso o cicatricial desorganizado incapaz de ejercer las funciones del hígado. Los hepatocitos pierden su arquitectura normal, y el lobulillo hepático se convierte en un conglomerado de células y tejido fibroso. Esta pérdida de estructura del lobulillo lo incapacita para realizar sus funciones (metabolización de sustancias, depósito de glucosa, producción de proteínas y factores de coagulación) por lo que el cuadro clínico de la cirrosis es muy amplio. FISIOPATOLOGIA DE LA CIRROSIS HEPÁTICA En el desarrollo de la fibrosis hepática y por lo tanto en la patogénesis, están involucrados los incrementos o modificaciones en la síntesis de colágeno y de otros componentes del tejido conjuntivo y de la membrana basal. La matriz extracelular en función celular se encuentra involucrada en la modulación de las actividades de las células con las cuales están en contacto, por tanto, la fibrosis puede afectar no sólo la física del flujo sanguíneo a través del hígado sino también la función de las células en sí mismas. La fibrosis hepática tiene lugar en 3 situaciones: 1) como respuesta inmunitaria vgr. VHB, 2) como parte de los procesos de cicatrización de las heridas vgr. VHA y tetracloruro de carbono, 3) en respuesta a agentes inductores de fibrogénesis primaria como el etanol y hierro. El responsable directo de todos los mecanismos de incremento en la fibrogénesis pueden ser las células almacenadoras de grasa del sistema retículoendotelial hepático. Estas células en respuesta a citocinas se diferencian de células quiescentes en las cuales se almacena la vitamina A en el interior de miofibroblastos que pierden la capacidad para almacenar la vitamina A y se incorporan activamente a la producción de matriz extracelular. Al parecer la fibrosis hepática tiene lugar en dos etapas. La primera se caracteriza por un cambio en la composición de la matriz extracelular de un colágeno sin enlaces cruzados y no formadora de fibrilla, a un colágeno más denso y sujeto a la formación de enlaces cruzados. En ésta etapa la lesión hepática todavía puede revertirse. La segunda involucra la formación de enlaces cruzados en la colágena subendotelial, la producción de las células mioepiteliales y la destrucción de la arquitectura hepática con la apariciónde nódulos en regeneración. Esta segunda etapa es irreversible. Aparte de los posibles efectos de la función del hepatocito, el aumento de la fibrosis modifica notablemente la naturaleza del flujo sanguíneo en el hígado y surgen complicaciones importantes. Alexandra Isla Cepeda
- 4. Hemorragia digestiva variceal La hemorragia digestiva por rotura de varices gastroesofágicas es una de las complicaciones más graves de la hipertensión portal, siendo responsable de aproximadamente un tercio de las muertes en pacientes cirróticos. Entre el 50 y 60% de los pacientes cirróticos poseen varices gastroesofágicas, y alrededor del 30% de estos experimentarán un episodio de sangrado en los dos primeros años de diagnóstico. Los principales determinantes del riesgo de sangrado en los pacientes cirróticos son el tamaño de la variz y el grado de disfunción hepática. El cese del sangrado puede ocurrir hasta en un 30% de los casos, pero la mortalidad asociada a cada episodio puede ser tan elevada como del 40%. Los casos de hemorragia persistente y recurrente (70%) se asocian a un peor pronóstico. En el paciente con hígado cirrótico hay un marcado aumento de la resistencia al flujo sanguíneo portal, factor determinante de la hipertensión portal. Junto al aumento de la resistencia hepática, en la cirrosis existe un estado de hiperemia secundario tanto a un estado de vasodilatación arteriolar, como a un aumento de la volemia. Lo que hace que exista un aumento de flujo hacia el territorio portal, factor que también contribuye a la elevación de la presión en dicho territorio. Las varices esofágicas se forman cuando el gradiente de presión portal está por encima de 12 mmHg. Existen distintas observaciones que están a favor de establecer en esta cifra como umbral para que se produzca la hemorragia. Distintos estudios demuestran que la hemorragia por varices no se produce si la presión es menor de 12 mmHg. Las varices suelen formarse en la unión gastroesofágica (zona de confluencia de la circulación venosa con el trayecto submucoso de los vasos). Factores que facilitan la formación de varices en esta zona: la ausencia de tejido de sostén, la presión negativa intratorácica y la existencia de venas perforantes que comunican las varices con las colaterales periesofágicas. Presiones varicosas superiores a 18 mmHg. se asocian a hemorragia persistente o a recidiva precoz, mientras que los enfermos con presiones inferiores a esta cifra tienen un bajo riesgo de hemorragia. Siguiendo a la Ley de Laplace, son las varices de gran tamaño y con presión elevada las que presentan un mayor riesgo de hemorragia. Juárez Aguirre Auracarolina
- 5. FACTORES DE RIESGO CLINICOS - Insuficiencia hepática avanzada - Consumo continuo de alcohol - Presión variceal mayor a 18mmhg - Tamaño de las varices VARICES GASTRICAS Cirrosis: 6% Prevalencia HTP no cirrótica: 19% debe investigarse la permeabilidad del eje espleno portal Valores Presión Portal Normal: 3 a 5 mm Hg Hipertensión Portal: > 5mm Hg Hipertensión Portal clínica: > 10mmHg Sangrado por varices: ≥ 12 mmHg TRATAMIENTO Durante un episodio agudo de sangrado por varices, el objetivo inmediato es conseguir la estabilidad hemodinámica del paciente y el cese de la hemorragia activa. Clásicamente se ha considerado que el esquema terapéutico de una hemorragia por varices debe de estar basado en los siguientes elementos: reposición de la volemia, profilaxis de las complicaciones y medidas encaminadas al control de la hemorragia Existen estudios que demuestran la eficacia similar de la somatostatina en relación a la vasopresina para el control de la hemorragia. De la misma forma, ha sido comparada la eficacia de la somatostatina con otros métodos para el control de la hemorragia, como son el taponamiento con balón o la escleroterapia, demostrándose una eficacia similar en ambos casos. Se ha comprobado que la administración de somatostatina durante cinco días producía una reducción en la incidencia de recidiva hemorrágica precoz. Todo ésto hace de la somatostatina un fármaco de primera línea para el control de la hemorragia por varices. Juárez Aguirre Auracarolina
- 6. HEMORRAGIA DIGESTIVA VARICEAL CAUSAS: Rotura de Varicesgastroesofágicas HTP 1/3 PACIENTESCIRROTICOS 50-60% + Resistenciadel flujosanguíneoportalFactor determinantede hipertensiónportal + De 12mmH g VARICESESOFÁGICAS Distintosestudios demostraronque la hemorragiapor varicesnose produce si tiene unapresión menora 12mmhg 30 % -> Episodiode sangrado Determinado Tamaño de la Varice Grado de disfunciónhepática Varices suelen formarse en la unión gastroesofágica(zonade confluencia de la circulación venosa con el trayecto submucoso de los vasos) Factoresde RiesgoClínicos: - InsuficienciaHepáticaAvanzada - Consumocontinuode Alcohol - PresiónVariceal +de 18mmHg - Tamaño de lasVarices VARICESGASTRICAS: 6% PACIENTESCON CIRROSIS 19% HTP NOCIRROTICA Juárez Aguirre Auracarolina
- 7. Factorespredisponentes - Ausenciade tejidode Sostén - Presiónnegativaintratorácica - Existenciade Venasperforantesque comunican lasvaricescon las colateralesperi esofágicas 30% -> cese de sangrado PersistenciayRecurrenciase asociaa un peorpronóstico Valor Normal de presión portal (3- 5mmhg) TTO: profilaxis Escleroterapia Ligadura por Bandas Juárez Aguirre Auracarolina
- 8. 2. FISIOPATOLOGÍADE LA HEPATITIS VIRAL B Y C La hepatitis hace referencia a la inflamación del hígado. Puede ser causada por virus hepatotrópicos que afectan de manera primordial las células hepáticas o hepatocitos, por mecanismos autoinmunitarios o reacciones por fármacos y toxinas, o bien ser secundaria a otros trastornos sitstémicos. Los virus que inducen enfermedad sistémica y pueden afectar al hígado incluyen al virus Epstein-Barr (mononucleosis infecciosa), capaz de generar hepatitis leve durante la fase aguda; el citomegalovirus, en particular en neonatos y personas con inmunosupresión; los herpesvirus, y los enterovirus. Los virus hepatotrópicos conocidos incluyen al virus de la hepatitis A (VHA), al virus de la hepatitis B (VHB), al virus δ asociado con el virus de la hepatitis B (VHD), al virus de la hepatitis C (VHC) y al virus de la hepatitis E (VHE). Si bien todos éstos pueden inducir hepatitis aguda, difieren en cuanto al modo de transmisión y período de incubación; el mecanismo, grado y cronicidad del daño hepático, y la capacidad para evolucionar al estado de portador. La presencia de antígenos virales y sus anticuerpos puede determinarse mediante pruebas de laboratorio. Los estudios epidemiológicos han indicado que algunos casos de hepatitis infecciosase deben a otros factores. Un agente viral similar al VHC se clonó e identificó como virus de la hepatitis G (VHG), también denominado GBV-C23. Se ha identificado evidencia de VHG en el 2% de los donadores de sangre en Estados Unidos24. Sin embargo, el VHG no se vincula con alguna hepatopatía o con exacerbaciones de la enfermedad hepática. A) VIRUS DE LA HEPATITIS B CARACTERISTICAS GENERALES ORDEN: caudovirales FAMILIA: hepadnaviridae GENERO: orthohepadnavirus ESPECIE: virus de la hepatitis B Virus de tipo ADN humano más pequeño que se conoce. Único virus de ADN que se replica con transcriptasa inversa de un ARN viral Tiene envoltura formada por una doble capa de lípidos sensible a detergentes El reservorio del virus de la hepatitis B es el hombre. Organotropismo estricto: hígado y en menor medida los riñones y el páncreas del ser humano y el chimpancé. Frecuencia de hepatopatía crónica: 10% Tiene cuatro serotipos principales (adr, adw, ayr, ayw). Existen ocho genotipos del virus (a-h) (según la variación en la secuencia de nucleótidos del genoma viral que tiene distribución geográfica distinta, son útiles para rastrear la evolución y la transmisión del virus. El virus de la hepatitis b es entre 50 y 100 veces más infeccioso que el VIH. LEIVA VELASQUEZ DARWIN
- 9. ESTRUCTURA: DEL VIRUS DE HEPATITIS B a) Una zona más externade 7 nm de anchura de composiciónlipoproteica se halla formado por: constituyentes de la membrana de retículo endoplásmico (RE) o del aparato de Golgi de los hepatocitos infectados. Embebida en la membrana está el antígeno de superficie (HBsAg) b) Una zona interna de 27 nm denominada núcleo o core, electro denso ligeramente hexagonal (nucleocápside), donde encierra Al genoma El ADN polimerasa viral VHB-P Proteínas del huésped. Parte de su DNA se encuentra como cadena incompleta de doble hélice, formando un círculo de aproximadamente 3.200 pares de bases; su cadena de DNA negativa está completa, en tanto que la positiva se extiende unas dos terceras partes ADN polimerasa ADN de cadena doble incompleta Lípidos de la membrana Antígeno de superficie (HBsAg ) grande Antígeno core (HBcAg) Antígeno de superficie (HBsAg) pequeño Antígeno e (HBeAg ) Antígeno de superficie (HBsAg) mediano NUCLEOCAPSIDE LEIVA VELASQUEZ DARWIN
- 10. MECANISMOS DE TRANSMISIÓN DEL VHB Existen tres formas fundamentales de transmisión de la hepatitis B: 1. Percutánea: contacto con sangre o productos sanguíneos, agujas contaminadas, jeringas, instrumentos, hemodiálisis, abuso de drogas intravenosas, cirugía oral, tatuajes, perforaciones (piercing), acupuntura. 2. Contacto íntimo o sexual. 3. Transmisión perinatal. Teniendo en cuenta estos mecanismos de transmisión, se recomienda investigar para hepatitis B a los siguientes grupos de riesgo: 1. Nacidos en áreas endémicas para hepatitis B 2. Sexo hombre con hombre 3. Trabajadores sexuales 4. Drogadictos intravenosos 5. Pacientes en hemodiálisis 7. VIH positivos 8. Embarazadas 9. Contacto familiar, íntimo o sexual con individuo HBV positivo. FISIOPATOLOGÍADE LA INFECCIÓN POR VHB El virus de la hepatitis B no es citopático, y la inflamación hepática depende de la respuesta inmune del individuo infectado. A mayor respuesta inmunológica, se va a presentar más inflamación hepática y la infección aguda es clínicamente más evidente. Hay más riesgo de insuficiencia y falla hepática porque la respuesta inmune lleva a la destrucción masiva de los hepatocitos infectados, pero a la vez hay mayor probabilidad de curación y control de la infección. Si la respuesta inmune es deficiente, se va a manifestar menos en su forma clínica ya que la necro inflamación será menor, pero el riesgo de cronicidad se aumentará, como sucede en coinfectados con VIH o en pacientes inmunosuprimidos (ej. hemodiálisis, quimioterapia). INMUNIDAD ANTE EL VHB Una particularidad del VHB es su escaso efecto citopático sobre el hepatocito, el virus no suele destruir a las células que infecta; sin embargo, es la respuesta inmune que se desencadena tras la infección y encargada de la eliminación del agente patógeno, la responsable de las principales manifestaciones de la enfermedad. Los principales efectores de la respuesta innata son las células naturales killer (NK), las células dendríticas, el sistema del complemento, así como citocinas y quimiocinas Activación de los macrófagos e incremento de la actividad microbicida. Aquí están implicadas distintas citocinas. El IFNγ es la principal citocina bactericida, dado que junto a la toxina del virus induce la producción macrofágica de TNFa, lo que activa a los macrófagos y facilita la eliminación del patógeno. Un modo indirecto de activar el macrófago es a través de la IL-12 que induce la producción de IFNγ por las células NK y los linfocitos T. El IFNa como el IFNγ aumentan la expresión de moléculas clase I y II del complejo mayor de histocompatibilidad (MHC), lo que facilita el reconocimiento de antígenos asociados al MHC por los linfocitos T citotóxicos (LTC). Este efecto puede ser considerado como parte de la inmunidad adaptativa mediada por células. Inhibición de la replicación viral. Es llevada a cabo fundamentalmente por los IFNs tipo 1 (IFNa, IFNß e IFNO), ya que inducen la síntesis de enzimas que bloquean la replicación viral. LEIVA VELASQUEZ DARWIN
- 11. ESQUEMA1 Una vez que se produce la infección viral, las CPA (sobre todo los linfocitos B) presentan en su superficie epítopes virales (epítopes del antígeno HBc son prominentes para linfocito T) y se desplazan desde el lugar de la infección a los órganos linfoides primarios donde presentan estos antígenos virales a las células B y T naïve, que se convierten en linfocitos B y T específicos de patógeno. Los linfocitos B activados son los responsables de la inmunidad humoral y producen anticuerpos neutralizantes capaces de unirse a las partículas virales circulantes y así limitar su expansión.Los antígenos virales son presentados asociados a moléculas MHC de clase II a los linfocitos T CD4. Tras la activación antigénica las CPA secretan IL-1, IL-6 y TNFa, los cuales inducen expresiónde receptores deIL-2 en las células T y secrecióndeotras citocinas que, asuvez, producen reclutamiento de otras células T con actividad efectora y reguladora. La activación de las células B por los linfocitos Th da lugar a la producción y secreción de anticuerpos neutralizantes capaces de unirse a las partículas virales circulantes y así limitar la expansión viral mientras que los linfocitos T citotóxicos CD8 (LTCs) ejercen su acción antiviral a través de la producción de citocinas y de mecanismos citolíticos directos que inducen la necrosis y la apoptosis celular . La histólisis directa se lleva a cabo mediante la liberación de gránulos citolíticos (como las perforinas y granocinas) que interactúan y destruyen la membrana de la célula infectada. Un aspecto clave es que los linfocitos T CD8 requieren de la ayuda de los linfocitos TCD4 para completar su diferenciación y así poder ejercer correctamente sus funciones. Por tanto, una respuesta vigorosa de los linfocitos T CD4 y CD8 conseguirá eliminar el patógeno viral mientras que una respuesta débil contribuirá a la persistencia del virus en el huésped. VIRUS HEPATOCITOS CELULAS ENDOTELIALES CELULAS DE KUPFFWER IFN Y IL-1 IFN A TNF-A ACTIVACION ENDOTELIALDE CELULAS DE KUPFFER INHIBICION DELA REPLICACION VIRAL QUIMIOCINAS AUMENTO DE ACTIVIDAD CITOLITICA CELULAS NKY LINFOCITOST CD8 INDUCCION MOLECULAR DE ADHESION CELULAR RECLUTAMIENTO DE LEUCOCITOS LEIVA VELASQUEZ DARWIN
- 12. VIRUS CELULA DENDRITICA CD4+ TH0 MACROFAGO IL-12 IL-6 IL-10 NK LINFOCITOCD4+ ACTIVADO INMUNIDAD INNATA CD4+ TH1 IL-12 IFNY CD8+ CD8+ CD8+ CD8+ IFNY TNFA CD4+ TH2 LB CEL. PLASMATICA ANTICUERPOS IL-4 IL-6 IL-5 IL-8 IL-13 2. RESPUESTA HUMORAL 1. RESPUESTA CELULAR INMUNIDAD ADAPTATIVAESQUEMA 2 LEIVA VELASQUEZ DARWIN
- 13. COMPLICACIONES DE LAINFECCIÓN CRÓNICAPOR VHB La infección crónica por VHB, cuando es progresiva, conlleva a complicaciones serias que comprometen la vida del individuo infectado, o que eventualmente va a obligar a someterlos a un trasplante hepático como última alternativa. Estas complicaciones se pueden dividir en dos grandes grupos: hepatocarcinoma y cirrosis. Hepatocarcinoma: cuando la infección se vuelve cró- nica, el genoma del virus B se incorpora al hepatocito, y puede causar mutaciones que dan origen a células hepáticas aberrantes que evolucionarán hacia el desarrollo de hepatocarcinoma. El VHB es considerado el segundo carcinógeno después del cigarrillo y es el responsable de la mayoría de hepatocarcinomas en África y Oriente, mientras el VHC lo es en el mundo occidental. No necesariamente tiene que haber cirrosis como ocurre con el virus C, y puede desarrollarse incluso en portadores inactivos del virus B. Cirrosis: la otra complicación de la infección crónica, la cirrosis, puede progresar a procesos como insuficiencia hepática e hipertensión portal, llegando a cumplir criterios para trasplante hepático. Los resultados iniciales de trasplante en VHB eran muy malos debido a la recurrencia con hepatitis severa fibrosante colestásica que llevaba a la muerte temprana o a retrasplante. Por un tiempo estos pacientes no fueron considerados candidatos a trasplante por muchos centros. Con el uso de HBIG desde la fase anhepática y por tiempo indefinido postrasplante, manteniendo niveles adecuados de anticuerpos antisuperficie (AntiHBs) se logró mejorar la sobrevida a niveles similares de los trasplantados por otras causas. El problema es el costo de esta inmunoprofilaxis. Actualmente se están dando análogos nucleósidos y nucleótidos pre y postrasplante con resultados promisorios para prevenir la reinfección y que eventualmente pueden reducir el costo por el uso de HBIG. Se ha usado la lamivudina pero tiene el inconveniente de desarrollar mutaciones resistentes. Ahora, con nuevos medicamentos como el adefovir y entecavir, el riesgo de estas mutaciones es muy bajo. Marcadores serológicos. Tres antígenos bien identificados se relacionan con el virus: un antígeno nuclear, el HBcAg, que se ubica en la nucleocápside; un transcrito polipeptídico más largo con regiones prenucleares y nucleares, que se designa HBeAg; y un antígeno de superficie, el HBsAg, que se localiza en la cubierta externa del virus. La región prenuclear dirige al polipéptido HBeAg hacia la sangre, en tanto que el HBcAG permanece en los hepatocitos para controlar el ensamblaje de los viriones nuevos. Los antígenos del VHB evocan la síntesis de anticuerpos específicos: los anti-HBs, los anti-HBc y los anti-HBe. Estos antígenos (el HBcAg no circula libre en la sangre) y sus anticuerpos sirven como marcadores serológicos para dar seguimiento a la evolución de la enfermedad . El HBsAg es el antígeno viral que, como rutina, se cuantifica en la sangre la mayoría de las veces. Aparece antes del desarrollo de los síntomas, alcanza un valor máximo durante la enfermedad clínica y luego disminuye hasta alcanzar niveles indetectables en 3 a 6 meses. Su persistencia después de los 6 meses señala una multiplicación viral persistente, la infectividad y el riesgo de desarrollar hepatitis crónica. El HBeAg aparece en el suero poco después del HBsAg, e implica la presencia de multiplicación viral activa. La IgM anti-HBc se vuelve detectable poco después del inicio de los síntomas, en forma concurrente al inicio de la elevación de las transaminasas séricas. En el transcurso de los meses, el anticuerpo tipo IgM es sustituido por IgG anti-HBc. El anti-HBe se vuelve detectable poco después de la desaparición del HBeAg, y su aparición señala el inicio de la resolución de la enfermedad aguda. La IgG anti-HBs, un anticuerpo específico contra el HBsAg, aparece en casitodas las personas una vez que se elimina este último. LEIVA VELASQUEZ DARWIN
- 14. El desarrollo de anti-HBs señala la recuperación de la infección por VHB, la carencia de infectividad y la protección contra la infección de VHB en el futuro. El anti-HBs es el anticuerpo que se encuentra en personas que tuvieron una inmunización exitosa contra el VHB. La presencia de ADN viral (ADN del VHB) en el suero es el indicador más certero de hepatitis B. Tiene presencia transitoria durante el período presintomático y por un período breve durante la enfermedad aguda. La presencia de la polimerasa del ADN, la enzima que sirve para la multiplicación del virus, suele ser transitoria, pero persiste durante años en individuos que son portadores crónicos y es un indicador de que la infectividad persiste. INTERPRETACION/ PERFIL SEROLOGICO HBsAg Anti-HBc Anti-HBs HBeAg Anti-HBe HBcIgM HB aguda, estado portador persistente - + - - - + HB crónica, estado portador persistente - + - - + + Convalecencia- infección en pasado reciente - + -/+ - + - Recuperado perdida anti- HBsAg - + - - - - Recuperado- infección en pasado reciente - + + - - - LEIVA VELASQUEZ DARWIN
- 15. B) VIRUS DE LA HEPATITIS C CIFRAS Y DATOS IMPORTANTES La hepatitis C es una enfermedad del hígado causada por el virus del mismo nombre; ese virus puede causar una infección, tanto aguda como crónica, cuya gravedad varía entre una dolencia leve que dura algunas semanas, y una enfermedad grave de por vida. Aproximadamente un 15-45% de las personas infectadas elimina el virus espontáneamente en un plazo de seis meses, sin necesidad de tratamiento alguno. El restante 55-85% de las personas desarrollará la infección crónica. De esas personas, el 15-30% correrá el riesgo de cirrosis hepática en un plazo de 20 años. El virus de la hepatitis C se transmite a través de la sangre, y las causas de infección más comunes son las prácticas de inyección poco seguras, la esterilización inapropiada de equipo médico en algunos entornos de atención sanitaria y la transfusión de sangre y productos sanguíneos sin analizar. En todo el mundo hay entre 130 y 150 millones de personas infectadas con el virus de la hepatitis C. Un número considerable de esas personas con infección crónica desarrollarán cirrosis o cáncer de hígado. Aproximadamente 500.000 personas mueren anualmente por enfermedades hepáticas relacionadas con la hepatitis C. Los antivíricos pueden curar aproximadamente el 90% de los casos de infección por el virus de la hepatitis C, lo que reduce el riesgo de muerte por cáncer de hígado y cirrosis, pero el acceso al diagnóstico y tratamiento es limitado. En la actualidad no existe ninguna vacuna contra la hepatitis C, pero la investigación en esa esfera continúa. CARACTERÍSTICAS a .- CLASIFICACIÓN: Familia: Flaviviridae Género: Hepacivirus b.- MORFOLOGIA: -Tamaño: 50nm -Genoma: ARN positivo de una cadena (8 genes). -Cápside icosaedro. -Envoltura: 2 glucoproteÍnas virales específicas (E1 y E2) -Las regiones hipervariables dentro de las glucoproteinas determinan mutaciones extensas y variabilidad anti genética. LEIVA VELASQUEZ DARWIN
- 16. ACTORES QUE INFLUYEN EN LAEVOLUCIÓN DE LAVHC • Factores genéticos: el segmento DR5 del HLA II, está asociado a una menor incidencia de cirrosis en pacientes infectados con VHC. • Factores naturales: edad, género, raza. • Consumo de Alcohol. • Tabaquismo. • Coinfeccion con otros virus: VIH, VHA, VHB, virus linfotrófico humano de Linfocito T. • Provoca 3 tipos de enfermedades: hepatitis aguda con resolución de la infección, infección crónica persistente y progresión rápida grave. ENVOLTURA VIRALARN VIRAL PROTEINASDE LA ENVOLTURA VIRAL PROTEINA DE LA CAPSIDE LEIVA VELASQUEZ DARWIN
- 17. TROPISMO VIRAL Casi no hay duda de que el HCV se replica en el hepatocito; sin embargo, su replicación también puede ocurrir en otras células como linfocitos T, linfocitos B y monocitos según lo atestigua la presencia de RNA viral específico de HCV en dichas células. Las variaciones en cantidad descritas respecto de esta replicación extrahepática pueden deberse a la presencia de cepas de HCV más linfotrópicas que otras, una característica que tal vez se relacione con mutaciones en la región 5’UTR. Asimismo el RNA del HCV se identifica en lesiones cutáneas de personas con crioglobulinemia y vasculitis relacionada con HCV, en biopsias renales de pacientes con glomerulonefritis membranoproliferativa vinculada con HCV y en saliva, semen, lágrimas, orina y líquido de ascitis. HEPATITIS AGUDA POR VHC El virus de la hepatitis C (VHC) entra primeramente al organismo ya sea por transfusión sanguínea o algún otro factor, cuando este virus llega a la sangre primeramente va a penetrar en el hepatocito infectándolo y a la vez para poder replicarse; esta infección va a promover la liberación de IFN-α por el hepatocito, el cual permite la formación de las proteínas de Complejo Mayor de Histocompatibilidad I y II ( MHC I Y MHC II), estas proteínas sirven para poder realizar la presentación de antígenos a los CD8+ ( MHC I) y a los CD4+ (MHC II), entonces inicia la expansión de los CD4, se va a producir la presentación de antígenos por los hepatocitos a los CD4, lo que va a permitir su diferenciación en Th1 y en Th2, las células Th1 liberan citoquinas, entre ellas el IFN-γ, el cual va a permitir la proliferación de los CD8, por otro lado las células Th2 liberan también citoquinas, entre ellas la IL6, el cual permite mayor secreción de anticuerpos por las células B; estos anticuerpos vas a neutralizar a los virus impidiendo su replicación al actuar sobre su región hipervariable. Con la proliferación de los CD8, los hepatocitos van a realizar la presentación de antígenos a estas células mediante MHC I, lo que provocará que los CD8 realicen su acción de citotoxicidad destruyendo a las células que se encuentran infectadas por este virus, completando así el ciclo de las defensas inmunes durante una hepatitis aguda causada por el VHC. El período de incubación de la hepatitis C puede variar entre 2 y 26 semanas, con un promedio de 7 semanas, y solo el 20% desarrollan un cuadro clínico de hepatitis aguda con ictericia, dolor en hipocondrio derecho, mialgias, vómito y fiebre, usualmente entre las 2 y 12 semanas posteriores a la infección. Estas manifestaciones clínicas son indistinguibles de las causadas por hepatitis A o B aguda. Algunos estudios han encontrado que el virus de la hepatitis C puede explicar entre el 12% y 16% de las hepatitis agudas en Estados Unidos. Otros estudios han mostrado que la infección aguda se resuelve de manera espontánea en el 15% al 25% de los infectados, quienes usualmente son de raza blanca, con ictericia y bajos niveles de viremia. En cuanto al 75% al 85% restante de los infectados en forma aguda, éstos desarrollan viremia persistente y la mayoría permanecen asintomáticos, aunque una gran parte de ellos empiezan a mostrar cambios bioquímicos e histológicos compatibles con la progresión de la enfermedad. La falla hepática es una presentación inusual; sin embargo, en un estudio realizado en Italia se encontró que la mortalidad por hepatitis C aguda entre 1995 y 2000 fue del 0,1%, comparada con el 0,01% causada por la hepatitis A, pero menor que la causada por LEIVA VELASQUEZ DARWIN
- 18. hepatitis B, del 0,4%. En nuestro medio ya se han descrito casos de hepatitis aguda por virus de la hepatitis C. CD8+ CTL CELL B CD8+ CTL HEPATO CITO CD4+ CELL TH CD4+ CELL TH EXPANSION CLONAL MHC- I MHC- II TCR TCR IFN-A TH1-CITOCINAS IFN Y TNF A EXPANSION CLONAL TH1- TH2 LISIS ACTIVACION Y DIFERENCIACION NEUTRALIZACI ON DE VHC POR ANTICUERPOS VHC ENTRADA VIRAL LEIVA VELASQUEZ DARWIN
- 19. HEPATITIS CRONICA En la hepatitis C crónica, los CD8 continúan produciendo su acción de citotoxicidad sobre los hepatocitos infectados, mientras los CD4 continúan produciendo citoquinas para mantener la respuesta inmune activa, manteniendo la proliferación y acción de CD8 para continuar evitando la propagación del VHC, sin embargo por el prolongado tiempo de infección del VHC, van a activarse las células de Kupffer del hígado, debido a su estimulación por el IFN- γ producido por los CD4 Th1, entonces estas células de Kupffer al activarse secretarán TGF- β, el TGF- β provoca en las células estrelladas del hígado activadas un aumento de la síntesis de las fibras colágenas, permitiendo la mayor producción de colágeno tipo I, que se depositan progresivamente en el espacio intercelular y en el espacio de Disse, lo que altera la función y la homeostasis de las células hepáticas, esta proteína está implicada en el desarrollo de la fibrosis hepática. La fibrosis progresiva en el parénquima hepático eventualmente causará cirrosis o pérdida de la función hepática. La mayoría de los pacientes con infección crónica son asintomáticos o solo presentan síntomas leves o inespecíficos. La queja más frecuente es la fatiga; otras manifestaciones menos comunes incluyen náuseas, anorexia, mialgias, artralgias, debilidad y pérdida de peso. Los síntomas rara vez son incapacitantes y pueden ser difíciles de atribuir a enfermedad hepática solamente y no a otras enfermedades como la depresión. Estos síntomas pueden ocasionar una disminución en la calidad de vida. Algunos pacientes infectados pueden desarrollar la enfermedad hepática severa después de pocos años, pero la mayoría la desarrollan luego de varias décadas; sin embargo, prácticamente todos los individuos infectados presentan anormalidades histológicas. CD8+ CD4+ CITOCINAS: IL-2,IFN Y,TNF,TGF-B,PDGF CEL. KUPFFER ACTIVACION DE FIBROBLASTOS TGF-B FIBROSIS MUERTE HEPATOCITOS PERFORINAS, GRANZIMAS LEIVA VELASQUEZ DARWIN
- 20. EVOLUCIÓN DE LA INFECCIÓN El paso de una infección aguda hasta una crónica se presenta sin aparición a síntomas, del 15% al 25% de los pacientes con hepatitis aguda pueden eliminar la infección, en tanto que del 75% al 85% de individuos afectados progresan a la fase crónica, de los infectados crónicamente el 20% desarrollan cirrosis hepática y de estos el 6% hace falla hepática terminal y entre el 1% y 4% desarrollan carcinoma hepatocelular. VIRUS DE LA HEPATITIS C INFECCION AGUDA CURACION Y ELIMINACION CIRROSIS DE INICIO RAPIDO INFECCION PERSISTENTE 15 % 15% 70 % ASINTOMATICOS HEPATITIS CRONICA INSUFICIENCIA HEPATICA CIRROSIS CARCINOMA HEPATOCELULAR 40% 6% 20% 4% LEIVA VELASQUEZ DARWIN
- 21. SINTOMATOLOGÍA La mayoría de los casos la infección se presenta de forma asintomática e anictérica. La infección crónica generalmente no presenta síntomas esta que el hígado llegue a un estado cirrótico. MARCADORES SEROLÓGICOS. Se dispone de pruebas de anticuerpos y virales para detectar la presencia de infección por VHC . Pueden obtenerse resultados negativos falsos en personas con inmunocompromiso y en una fase temprana de la evolución de la enfermedad, antes de que se desarrollen anticuerpos. La cuantificación directa del VHC en el suero sigue siendo la prueba más precisa para identificar la infección. Las pruebas virales son muy sensibles y específicas, pero más costosas que las de anticuerpos. Con las técnicas más recientes para análisis de anticuerpos, a menudo la infección puede detectarse incluso entre 6 y 8 semanas después de la exposición, o bien en 1 o 2 semanas con las pruebas virales que recurren a las técnicas de polimerasa de reacción en cadena. A diferencia de la hepatitis B, los anticuerpos contra el VHC no son protectores, pero sirven como marcadores de la enfermedad. LEIVA VELASQUEZ DARWIN
- 22. COLEDOCOLITIASIS Introducción La coledocolitiasis se refiere a los cálculos que son precipitaciones de cristales, formadas tanto de colesterol como cálculos pigmentados, estos se encuentran en los conductos biliares. Existen 2 tipos: Primaria: Es rara y se da por formación de piedras dentro de los mismos conductos biliares Secundaria: Es la más frecuente y se debe a piedras que se forman en la vesícula y salen de esta para impactarse en los conductos biliares La formación de los cálculos en la vesícula biliar, cuando la bilis esta sobresaturada con colesterol, las micelas mixtas son insuficientes para solubilizarlo, por lo que abundan vesículas unilamelares (liposomas) para solubilizar el colesterol Estas vesículas pueden fusionarse para convertirse en multilamelares, en donde el colesterol cristaliza La producción de lodo biliar, en su mayoría mucinas y la escasa motilidad vesicular favorecen la formación del cálculo, que puede crecer y generar sintomatología La presión intracoledociana es normalmente de 10 a 15 cm de agua, cuando la presión excede 15 cm de agua el flujo biliar disminuye y cuando llega a 30 cm de agua se detiene, la presión de un colédoco obstruido puede alcanzar hasta 40 cm de agua. LINARES LLATAS LHESLY
- 23. COLEDOCOLITIASIS Se define comolapresenciade cálculos(precipitacionesde cristales de colesterol ode bilirrubinaque se desarrollanenel interiorde lavesícula biliar) enlosconductosbiliares. Coledocolitiasisprimaria Es rara y se da por formaciónde piedrasdentrode losmismos conductosbiliarescomune incluso dentrodel hígadoen personascon “bilislitogenica” Coledocolitiasissecundaria Es la más frecuente yse debe a piedrasque se formanenla vesículay salende estapara impactarse enlos conductosbiliares Manifestaciones Clinicas Clasificación Diagnostico Signosy síntomas: Ictericia Coluria Colangitis Fiebre Escalofríos Nauseas Vomitos Doloren laparte superior derechao central del abdomen Acolia El principal estudioparasu diagnósticoesel ultrasonidoy las pruebasde funciónhepática Se realizaratambién: - Colangioresonanciamagnética(diagnóstico diferencial) - Colangiografíaretrogradapancreática endoscópica(CPRE) permitelalimpiezadel conductobiliar,laextracciónde laspiedras - Esfinterotomíaparapermitirque labilis fluyacon facilidad. LINARES LLATAS LHESLY
- 24. FISIOPATOLOGIA Insuficiencia en la producción de quenoxedoico Descensoenla secreción de sales biliares y lectina Exceso en la producción o excrecion de colesterol Aumento de colesterol Desconjugacion de la bilirrubina conjugada con la bilis Excesoen la bilirrubina libre Cálculos pigmentadosCalculos de colesterol Interaccion con calcio y cobre, ayudan a precipitar los cálculos pigmentados de bilirrubina Bilis sobresaturada de colesterol Concentración de acidos biliares y fosfolipidos Secreciónde colesterol no esterificado Mucina Factoresantinucleares Motilidad vesicular vesicular Microcristalesde colesterol monohidratado Formación de calculos LINARES LLATAS LHESLY
- 25. Cálculos biliares Irritación por acidos biliares Inflamación de la mucosa Exposición del epitelio Destrucción de la mucosa Paso de sales biliares en los tejidos Obstrucción del flujo biliar Presión intraluminal Distención vesicular Obstruccion linfática y venosa Isquemia y necrosis LINARES LLATAS LHESLY
- 26. TOXINA COLERICA La especie no es homogénea en lo que respecta a su potencial patógeno. La presencia de antígenos somáticos termoestables O, ha permitido determinar más de 200 serogrupos de V. cholerae. Solamente dos de ellos se reconocen como responsables de las epidemias de Cólera, el O1 y el O139. El genoma consiste en dos cromosomas circulares de 2.961.146 pb (cromosoma 1) y 1.072.314 pb (cromosoma 2). La mayoría de los genes esenciales para la patogenicidad, como la toxina colérica, el pilus co-regulado con toxina, los lipolisacáridos y la maquinaria de secreción de proteínas extracelulares, están localizados en el cromosoma 1. La enterotoxina de V. cholerae está codificada por los genes ctx. El gen ctxA codifica la subunidad A de la toxina, y el gen ctxB codifica la subunidad B. Los genes son parte del mismo operón ctxAB. El transcrito (ARNm) del operón ctx tiene dos sitios de unión a ribosoma (RBS), uno en el gen A y otro en el gen B. Este último es, por lo menos, siete veces más fuerte que el sitio de la región A. De esta forma el organismo es capaz de traducir más proteínas B que A, lo cual se requiere para ensamblar la toxina en la proporción apropiada (1A:5B). Los componentes son ensamblados en el periplasma después de la traducción. Cualquier subunidad B extra puede ser excretada por la célula, pero la subunidad A debe estar adherida a 5 subunidades B, para poder salir de la célula. La subunidad A intacta no es activa enzimáticamente, y se debe fraccionar en los fragmentos A1 y A2, los cuales están unidos por un puente disulfuro. La diarrea masiva inducida por V. cholerae es causada por la toxina colérica. Tras penetrar por vía oral y sobrepasar la barrera gástrica, los vibriones se multiplican en el intestino y tienden a fijarse en la mucosa produciendo una mucinasa que hidroliza el moco intestinal, la hemaglutinina, que fragmenta la fibronectina, contribuyendo a la activación proteolítica de la toxina colérica. Esta proteasa facilita la debilitación del moco, el cual se produce en la superficie intestinal y contribuye a despegar los protectores del receptor para las adhesinas. Cada molécula de la toxina colérica, está compuesta por cinco subunidades B (de enlace) y una subunidad A (activa). Las subunidades B se unen a los receptores del gangliósido GM1 en las células epiteliales de la mucosa intestinal. Después de la unión, se separan la subunidad A1 y el componente A2, lo cual facilita la entrada del componente A1 en la célula. El componente A1 de la toxina colérica estimula la producción de la enzima adenilciclasa, involucrada en la producción del monofosfato cíclico de adenosina (AMPc). El AMPc intracelular estimula el canal de cloro en las criptas intestinales, lo que incrementa la secreción de agua y electrólitos e inhibe el cotransporte de sodio y cloro en las células de las vellosidades. Como resultado de estas 2 acciones en las criptas y en las vellosidades por la toxina colérica, la secreción de líquidos en el lumen intestinal lleva a una diarrea acuosa con una concentración de electrolitos similar a la plasmática. A parte de la toxina colérica, el efecto patógeno del V. cholerae se incrementa a través de las fimbrias que facilitan una mayor adherencia a las mucosas, la adhesina para conseguir
- 27. una colonización accesoria, diversas sustancias como mucinasa o proteasa que provocan inflamación intestinal o la neuraminidasa que aumenta los receptores de la toxina. No obstante, el V. cholerae no invade la pared intestinal y se observan pocos neutrófilos en las heces. En la forma grave, el cuadro clínico clásico, aparece bruscamente diarrea acuosa, con deposiciones típicamente poco dolorosas y muy abundantes (pueden alcanzar los 500- 1.000 ml/hora), sin tenesmo, con característico aspecto de agua de arroz y olor a pescado. Pueden presentarse vómitos no precedidos de náuseas abundantes, acuosos y alcalinos. Suelen aparecer con posterioridad a la diarrea. Todo ello produce en horas una grave deshidratación con oliguria y calambres musculares, que pone en peligro la vida del paciente. Inicialmente, aparece sed intensa e intranquilidad, para evolucionar a apatía y shock hasta desembocar en coma. Puede aparecer en algunos casos acidosis con aumento del anión gap, secundaria a las pérdidas de bicarbonato por heces y a la acidosis láctica, con descenso del pH y del bicarbonato. Esta acidosis se asocia a cifras de potasio normales o altas, a pesar de la intensa pérdida de este ión.
- 28. SALMONELLA La presencia de cápsula y flagelo en Salmonella depende de la especie en cuestión, solamente S. enterica serovar Typhi, S. enterica serovar Paratyphi C y S. enterica serovar Dublin presentan cápsula y todas las salmonelas se consideran móviles a excepción de S. enterica serovar Gallinarum. E. Esta bacteria expresa una amplia variedad de fimbrias con diferente especificidad de unión Las salmonellas son bacterias invasoras que penetran por la vía oral, deben superar varias barreras o líneas de defensa naturales para finalmente localizarse en el ileon terminal y el colon proximal. La enfermedad va a depender fundamentalmente de la cantidad de microorganismos ingeridos (inóculo), de su virulencia y de factores dependientes del huésped. Se precisa, por término medio, un inóculo superior al millón de gérmenes. La acidez gástrica es una barrera natural importante, siendo factores predisponentes aquellas circunstancias que modifican el pH gástrico, como aclorhidria, vagotomía, gastrectomía o la toma de fármacos que lo modifican. Por otro lado, las salmonellas tienen capacidad de adaptarse a ésta y pueden sobrevivir a pH de 4,0. Después de pasar el estómago las salmonellas llegan al intestino delgado donde interactúan con las células de la pared intestinal. Los péptidos catiónicos antibacterianos, secretados por las células de Paneth del intestino delgado podrían representar una importante segunda línea de defensa contra salmonella y otros patógenos. Se adhiere apicalmente a las células epiteliales del íleon y a las células M que debido a la ausencia del borde de cepillo así como de glicocalix, representan una puerta de entrada ideal para las enterobacterias. La capacidad de la Salmonella para sobrevivir dentro de los macrófagos probablemente es esencial en la patogenia de la fiebre tifoidea y en la diseminación de los gérmenes a la circulación sistémica a través del conducto torácico. Finalmente los gérmenes son captados por los macrófagos tisulares en la medula ósea, el bazo y las placas de Peyer. Las placas de Peyer se muestran tumefactas pudiéndose ulcerar la mucosa intestinal pasada la primera semana y originar una hemorragia o la perforación, las dos complicaciones más graves del cuadro. Los mecanismos de patogenicidad con que Salmonella induce diarrea y septicemia no han sido descritos detalladamente, pero parece ser un fenómeno complejo que involucra diversos factores de virulencia. Se ha demostrado la presencia de enterotoxina en S. enterica serovar Typhimurium y S. enterica serovar Typhi similar a las enterotoxina de Vibrio cholerae (CT) y toxina termolábil (LT) de E. coli. Salmonella induce la migración de neutrófilos y macrófagos (PMN) así la liberación de citocinas proinflamatorias como IL8, GM-CSF, IFNγ, TNFα, GCP-2 (proteína 2 quimiotáctica de granulocitos) GRO-α (gen α relacionado a crecimiento), GRO-β y GRO- γ que reclutan células fagocíticas y están involucradas en el proceso diarreico, también se habla de un quimioatrayente, aún no caracterizado, conocido como PEEC (pathogen- elicited epithelial chemoattractant).
- 29. En la cara apical y lateral de los enterocitos sobreexpresa ICAM-1, que colabora en el movimiento fagocítico de los neutrófilos. La producción de citocinas proinflamatorias inducida por Salmonella es debida a la activación de factores de transcripción NF-kB y AP-1, como consecuencia de la estimulación de las MAP cinasas por parte de la proteína efectora SopE; SopB activa Akt, quinasa serinatreonina que puede regular la actividad transcripcional de NF-kB. El incremento de la permeabilidad vascular que acompaña la inflamación en combinación con la perdida de la integridad epitelial de la mucosa intestinal provoca la diarrea, al ocasionar un desequilibrio entre la secreción y la absorción de líquidos incrementando la salida de líquidos y electrolitos al lumen intestinal.
- 30. Vibrio Cholerae Patogenicidaddadaporunaenterotoxina TOXINA COLERICA Gen localizadoenel Cromosoma1 compuesta por 5 subunidadesB (de enlace) SubunidadA (activa) Receptores gangliosidosGM1 En célulasepiteliales de la mucosa intestinal CodificadoporgenctxB CodificadoporgenctxA Ensambladosen el periplasma Más de 200 serogrupos. Responsables de las epidemias de Cólera, el O1 y el O139. Genoma consiste en dos cromosomas circulares de 2.961.146 pb (cromosoma 1) y 1.072.314 pb (cromosoma 2). Estimulaproducciónde enzimaadenil ciclasa La subunidad A debe estar adherida a 5 subunidades B, para poder salir de la célula para poder salir de la célula.
- 31. VIBRIO CHOLERAE Penetra por la vía oral y sobrepasa la barrera gástrica. En el Intestino delgado Vibriones se multiplican Mucinasa hidroliza moco intestinal Hematoglutinina, fragmenta la fibronectina Se despegan los protectores del receptor para adhesinas Las subunidades B se unen a los receptores del gangliósido GM1 El componente A1 de la toxina colérica estimula la producción de la enzima adenilciclasa Producción del monofosfato cíclico de adenosina (AMPc) Estimula el canal de cloro en las criptas intestinales Incrementa la secreción de agua y electrólitos Inhibe el cotransporte de sodio y cloro en las células de las vellosidades TOXINA COLERICA Secreción de líquidos en el lumen intestinal lleva a una diarrea acuosa con una concentración de electrolitos similar a la plasmática.
- 32. Cuadro clinico clasico Diarrea acuosa Deposiciones poco dolorosas 500-1000 ml/hora Vomitos Polidipsia Deshidratación con oliguria y calambres musculares Acidosis con aumento del anión gap Descenso del pH y del bicarbonato. Cifras de potasio normales o altas
- 33. SALMONELLA Bacterias invasoras que penetran por la vía oral. Después de pasar el estómago las salmonellas llegan al intestino delgado donde interactúan con las células de la pared intestinal. Proceso inflamatorio Incremetno de la permeabilidad vascularPerdida de la integridad epitelial de la mucosa intestinal Desequilibrio entre secreción y absorción de líquidos incrementando la salida de líquidos y electrolitos al lumen intestinal.
- 34. SALMONELLA Migración de Neutrófilos y Macrófagos Liberación de citosinas pro inlfamoatorias como Il-8, GM-CSF, IFNγ, TNFα, GCP-2 Reclutan células fagocíticas Sobreexpresa ICAM-1, en la cara apical y lateral de los enterocitos. Involucrado en el proceso diarreico. Activación de factores de transcripción NF-kB y AP-1 debido a
- 35. DIARREA AGUDA INFECCIOSA POR ESCHERICHIA COLI Escherichia Coli Características • Bacilo gram negative no forma esporas • Movilesmiden 5 u de ancho por 3 u de largo • Catalase positivos • Oxidase negativos • Producen vitamin B Y K ESTRUCTURA ANTIGENICA • Antígeno capsular ( antígeno ¨K¨) • Antígeno somatico ( ANTIGENO ¨O¨) • Antígeno flagelar (ANTIGENO ¨H¨) • Antigenos menores como proteínas de membrana externa y fimbrias DIARREA AGUDA INFECCIOSA Definición LIZARBE ASMAT, PAOLA ARACELY
- 36. Enfermedad diarreica aguda es el término adoptado por la Organización Mundial de la Salud (OMS) para referirse a un proceso inflamatorio gastrointestinal infeccioso o no infeccioso, asociado a una disminución en la consistencia y un aumento en la frecuencia ( > 3 veces por día) de las deposiciones fecales. La diarrea puede ser líquida, semilíquida, y puede contener moco y sangre. Otras manifestaciones clínicas de la EDA son náuseas, vómitos, dolor abdominal, fiebre y deshidratación. Como en algunos pacientes el vómito, la deshidratación, y la fiebre pueden ser las manifestaciones más predominantes, un término apropiado para describir los órganos afectados y las manifestaciones clínicas de los pacientes afectados sería el de gastroenteritis aguda. Teniendo en cuenta que EDA es el término adoptado por la OMS, y por el Ministerio de Protección Social de la República de Colombia, EDA será el término que se usará a todo lo largo de esta revisión. La diarrea aguda casi siempre se debe a infección. Estas infecciones se adquieren a su vez en la mayoría de las ocasiones a través del agua, de los alimentes, por la via fecal-oral, y persona a persona como ocurre en las toxinfecciones alimentarias y en la diarrea del viajero. Otrs veces es por via aérea como en las gastroenteritis viral flora intestinal, los que favorecen la aparición de cuadros infecciosos intestinales como ocurre en la diarrea secundaria. Causas, incidencia y factores de riesgo La E.coli es un tipo de bacteria que normalmente vive en los intestinos de los humanos y los animales sin causar ningún problema. Sin embargo, ciertos tipos (o cepas) de E. coli pueden ocasionar intoxicación alimentaria. Una cepa (E. coli O157:H7) puede provocar un caso grave de este tipo de intoxicación. La bacteria puede ingresar al alimento de diferentes maneras: La carne de res o de aves puede entrar en contacto con las bacterias intestinales cuando la estén procesando. El aguaque seutiliza durante elcultivo o embarque puede contener desechos humanos o animales. Manipulación o preparación inapropiada de los alimentos. La intoxicación alimentaria con frecuencia ocurre por comer o beber: Cualquier alimento preparado por alguien que no se lavó las manos apropiadamente. Cualquier alimento preparado usando utensilios de cocina, tablas de cortar y otras herramientas sucios. Productos lácteos o alimentos que contengan mayonesa (como ensalada de col o de papas) que hayan permanecido por fuera del refrigerador por mucho tiempo. Alimentos congelados o refrigerados que no se guarden a la temperatura apropiada o que no se recalienten adecuadamente. Pescados u ostras crudas. Frutas o verduras crudas que no se hayan lavado bien. LIZARBE ASMAT, PAOLA ARACELY
- 37. Jugos de verduras o frutas crudas y productos lácteos. Carnes o huevos mal cocidos. Agua proveniente de un pozo o arroyo, o agua de una ciudad o pueblo que no haya sido tratada. Aunque no es común, la E. coli se puede diseminar de una persona a otra. Esto puede suceder cuando alguien no se lava las manos después de una defecación y luego toca otros objetos o las manos de otra persona. MECANISMOS PATOGÉNICOS El mecanismo patogénico que ocasiona la diarrea es variable, y está en dependencia del grupo o categoría de E. coli que infecte al individuo. Así podrá existir: • Invasividad. • Adherencia a la superficie de la mucosa. • Producción de enterotoxinas. • Producción de citotoxinas. FACTORES DE VIRULENCIA ESPECIALIZADOS ASOCIADOS A ESCHERICHIA COLI BACTERIA ADHESINA EXOTOXINAS ECET Antigenos del factor de colonización ( CFA/I, CFA/II, CFA/III) Toxina termolábil (LT-1); toxina termoestable (Sta) ECEP Pili formadores de haces ( BFP); intimina ECEA Fimbria adherentes agregantes ( AAF/I, AAF/II, AAF/III) Toxina termoestable enteroagregante; toxina codificada por plásmidos ECEH BFP; Intimina Toxinas de Shiga (Stx-1, Stx-2) ECEI Antígeno del plásmido invasivo Hemolisina (Hly A) LIZARBE ASMAT, PAOLA ARACELY
- 38. CLASIFICACIÓN E. COLI ENTEROTOXIGÉNICA Es el agente más frecuentemente asociado a EDA infantil en el mundo y el que está asociado con mayor mortalidad y morbilidad en niños bajo 5 años de edad, después de rotavirus22. Además, se le reconoce como el agente más frecuentemente asociadoadiarrea del viajero23. ECET se define como aquellas cepas de E. coli que expresan la enterotoxina termolábil LT y/o la enterotoxina termoestable ST y que son capaces de colonizar el intestino humano mediante factores de colonización24. Más de 22 factores de colonización constituidos por pili y adhesinas no asociadas a pili permiten la adherencia bacteriana a células intestinales, facilitando la colonización del intestino delgado LT es una toxina conformada por una subunidad A y cinco subunidades B. La subunidad A activa permanentemente la enzima adenilatociclasa que al elevar los niveles de AMP-cíclico intracelular, inducen la fosforilación del regulador de conductancia transmembrana de la fibrosis quística (CFTR) y su apertura. La apertura de este regulador resulta en secreción activa de cloro y agua de células intestinales, lo que se traduce en una diarrea secretora intensa. El mecanismo de acción de la LT es casi idéntico al de la toxina colérica (CTX) de V. cholerae. La subunidad B se une específicamente al receptor gangliosido M1 sobre lasuperficie apicalde células intestinales,através del cual la subunidad A entra al citoplasma a activar la adenilatociclasa LIZARBE ASMAT, PAOLA ARACELY
- 39. E. COLI ENTEROINVASIVA Induce una diarrea disentérica muy semejante a Shigella spp. aunque también se asocia a diarrea acuosa y afecta a niños, principalmente en países de bajos recursos49. Estudios filogenéticos de ARN ribosomal y de análisis de ADN de los plásmidos de virulencia indican que ECEI es un ancestro cercano a la Shigella50. Dicho plásmido de virulencia codifica un sistema de secreción tipo III similar en estructura al de ECEP pero su función está dirigida a la inducción de invasión bacteriana de las células intestinales y su diseminación intercelular. Este fenotipo caracterizado por destrucción celular explica el síndrome disentérico en el paciente afectado. E. COLI ENTEROHEMORRÁGICA Contagio por alimentos contaminados Baja dosis infectiva (<100 bacterias) Causa desde diarreas leves hasta colítis hemorrágica Toxinas VT1 y VT2 codificadas en dos profagos. Parecidas a las tóxinas de Shigella Puede producir SHU por daño renal posterior a las colítis LIZARBE ASMAT, PAOLA ARACELY
- 40. E. COLI ENTEROADHERENTE O ENTEROAGREGATIVA Se ha descrito también como un agente importante de diarrea del viajero. ECEA se define como aquella E. coli que induce adherencia agregativa en células epiteliales, mediada por la fimbria AFF. La adherencia agregativa sobre las superficies celulares o no celulares semeja el patrón de ladrillos de pared. Este tipo de adherencia favorece no sólo la colonización intestinal por este pato-tipo sino que también la formación de biopelículas sobre superficies no biológicas. E. COLI ENTEROPATOGÉNICA Contagio por alimentos y aguas contaminadas La bacteria se adhiere al epitelio del intestino y destruye las microvellosidades Los factores de virulencia están codificados en una “isla de patogenicidad” y plasmidio La diarrea se produce generalmente en niños pequeños, por la reducción de la absorción LIZARBE ASMAT, PAOLA ARACELY
- 41. DIARREA AGUDA INFECCIOSA POR ESCHERICHIA COLI ESCHERICHIA COLI Causa importante de mortalidad en niños menores de 5 años. .La infección es más frecuente durante los meses de verano o época de lluvias en las regiones tropicales. La diseminación de las E. coli diarreogénicas es por contaminación fecal de agua o alimentos. Principales agente etiológico de la diarrea del viajero. Frecuentes en el tubo digestivo Bacilos Gram negativos Anaerobiofacultativos Fermentadores; oxidasa-negativo Lipopolisacárido consta de un polisacárido externo somático O, un núcleo polisacárido( antígeno común) y lípido A ( endotoxina) Duración <2 semanas. Evacuaciones>3/24h. Comienzobrusco. Dolorabdominal difuso. Dolorperi umbilical. Consistencialíquidas,pastosas. Autolimitadas Epidemiología. Manifestaciones Clínicas Los síntomas se desarrollan de 24 a 72 horas después de resultar infectado. Fiebre Gases Inapetencia Cólicos estomacales Vómitos (raro) Los síntomas de una infección por E. coli infrecuente pero grave abarcan: Hematomas que se presentan fácilmente Piel pálida Orina roja o con sangre Disminución de la cantidad de orina La diarrea que es súbita, intensa y a menudo con sangre es el síntoma más común. Clasificación E. coli enteropatógena (ECEP) E. coli enterohemorrágica (ECEH) E. coli enteroagregativa (ECEA) E. coli enterotoxigénica (ECET) E. coli enteroinvasiva (ECEI) Emisión de heces líquidas de consistencia disminuida y va acompañado por el aumento de frecuencia del ritmo deposicional. LIZARBE ASMAT, PAOLA ARACELY
- 42. Abrev. NOMBRE E. COLI LUGAR DE ACCIÓN POBLACIÓN EN RIESGO ENFERMEDAD PATOGENIA DURACIÓN PRINCIPALES FACTORES DE VIRULENCIA ECET Enterotoxigéncia Intestino delgano > 1 año Viajeros Diarrea del viajero, diarrea infantil enpaísesendesarrollo; diarrea acuosa, vómitos, espasmos abdominales, náuseas, febrícula Enterotoxina termoestable y/o termolábilesmediadasporplásmidos que estimulan la hipersecreción de liquidos y electrólitos Aguda LT, ST, CFA ECEP Enteropatogénica Intestino delgano 2 años Diarrea infantil en países en desarrollo; diarrea acuosa y vómitos; heces no sanguinolientas. Histopatologia A/B mediada por plásmidos con la alteración de la estructura normal de la microvellosidad, lo que da lugar a la mala absorción y diarrea Aguda Persistente Eae, bfp ECEA Enteroagregativa Intestino delgano > 1 año Viajeros Pacientes VIH Diarrea infantile en países en desarrollo y desarrollados; diarrea del viajero, diarrea acuosa persistente con vómitos, deshidratación y febrícula. Adherencia agregativa de los bacilos mediada por plásmido ( ¨ladrillos apilados¨) con acortamiento de las microvellosidades, infiltración mononuclear y hemorragía; disminución de la absorción de líquidos Aguda Persistente AAF, dispesina EAST1 ECEH Enterohemorrágica Intestino grueso 6 meses – 10 años Ancianos Inicialmente diarrea aguosa seguida de diarrea sanguinolienta ( colitis hemorragica) con espamos abdominales, sin fiebre o con febricula; puede progresar a síndrome hemolítico urémico ECEH evolucionaapartir de ECEP; lesiones A/Bcon destrucción de la microvellosidad intestinal, que da lugar a disminución de la absorción; patología mediada por toxinas citotóxicas Shiga ( Stx-1, Stx-2), que interrumpen la síntesis de proteínas Aguda Stx1, Stx2, eae, hlya ECEI Eteroinvasiva Intestino grueso > 1 año Rara en lospaisesde desarrollo y desarrollados; fiebre, espasmos, diarrea acuosa; puede progresar a disentería con escasas heces sanguinolentas. Invasion mediada por plásmidos y destrucciónde lascelulasque recubren el colon Aguda Ipah, enterotoxina de Shigella LIZARBE ASMAT, PAOLA ARACELY
- 43. TUBERCULOSIS PERITONEAL E INTESTINAL La tuberculosis es considerada como la primera amenaza para la salud pública de la década debido a que en el mundo actual mueren más enfermos por tuberculosis que por cualquier otra enfermedad y no hace distingos de sexo, edad o situación económica. Por ello, en la Conferencia Mundial sobre Tuberculosis y Desarrollo Sostenible realizada en marzo del año 2000, en Ámsterdam, Holanda, representantes de las naciones y de la Organización Mundial de la Salud (OMS) expresaron la urgencia de tomar acciones inmediatas contra esta enfermedad endémic a que causa muerte en poblaciones como la nuestra y que incide en forma negativa en el desarrollo de los países. Durante la Asamblea Mundial de la Salud del año 2001, la Dra. Gro Harlem Brundeland, Directora General de la OMS, informó que el Perú ha salido de la lista de los 22 países que concentran el 80% de los casos estimados de tuberculosis en el mundo, habiéndose evitado numerosas muertes. La tuberculosis está incrementándose tanto en los países desarrollados como en los países en vía de desarrollo debido a tratamiento inadecuado de los enfermos, a la epidemia del VIH Sida, a la resistencia a los fármacos antituberculosos, así como al deterioro de las condiciones socioeconómicas, aumento de la pobreza y débil apoyo político y económico. Actualmente se considera que un tercio de la poblac ión humana se halla infectada por Mycobacterium tuberculosis. La tuberculosis es una enfermedad que puede comprometer diferentes aparatos y sistemas, con predominio del compromiso pulmonar en una relación de diez a uno sobre el extrapulmonar. El compromiso digestivo se encuentra entre 0,4% y 5% y en nuestro medio es predominanteme nte secundario a la existencia de un foco pulmonar, entre el 75% y 92,6%. Son raros los casos de compromiso primario por ingestión de leche recién extraída o no hervida12. En el Perú ha sido descrito el hallazgo de bacilos acidorresistentes en los pulmones y en los ganglios mesentéricos de momias que datan del año 700 d. C. En 1994 fue reportado el hallazgo de ADN de M. tuberculosis en una momia precolombina de Perú. El primer caso documentado de tuberculosis peritoneal data de 1843, en un hospital neoyorquino. Conocida como “La gran simuladora", la tuberculosis mimetiza toda una variedad de padecimientos. La tuberculosis todavía es un flagelo en los países en vías de desarrollo y un serio problema en algunas comunidades desarrolladas por lo que el interés en la enfermedad de localización abdominal es tema de actualidad. Durante el año 2000, Lima y Callao reportaron el mayor número de casos de tuberculosis (todas las formas) en relación con los demás departamentos del Perú (Programa Nacional de Control de la tuberculosis - Informe 2000, MINSA). Seis departamentos tienen tasas superiores a 150 x 100 000 habitantes. En ellos se concentra el 38 % de la población y se notifica el 63,4 % de los casos. Más del 67% de la población vive en zonas urbanas y tiene alta densidad poblacional. Según este informe, el factor más importante asociado con el riesgo de infección es el contacto próximo con pacientes con tuberculosis pulmonar bacteriológicamente positivos. Por lo que a mayor concentración y mayor prevalencia de tuberculosis pulmonar, mayores son las probabilidades de transmisión por vía aérea de los bacilos de la tuberculosis. A partir del año 2001, con la introducción de la información en el SYSTB, será posible obtener información sobre el número de casos por distrito y, además, permitirá identificar los “bolsones ALUMNO: ElvisLujánRuiz
- 44. de tuberculosis" con la finalidad de efectuar intervenciones específicas que lleven a lograr un mayor impacto en las acciones del control de la tuberculosis. EPIDEMIOLOGÍA La tuberculosis del aparato digestivo es una enfermedad asociada con la pobreza y a inadecuados sistemas de salud que predomina en personas adultas con antecedente de tuberculosis pulmonar o con tuberculosis pulmonar activa asociada a otras condiciones como infección por virus de la inmunodeficiencia (VIH), desnutrición, etilismo y/o drogadicción. El 95% de las muertes se produce en los países en vía de desarrollo. Es más frecuente en adultos jóvenes, el grupo de edad frecuentemente comprometido se encuentra entre la segunda y cuarta década de la vida con ligero predominio del sexo femenino sobre el masculino. En el Perú la proyección de la población total para el año 2000 ascendió a 25 661690 habitantes y se han diagnosticado y tratado a 39 918 personas enfermas con tuberculosis (en todas sus formas) en todo el país2. De todos los casos de tuberculosis, 86% correspondió a los nunca tratados y 14% a los anteriormente tratados. Asimismo, 22 580 personas fueron diagnosticadas de tuberculosis pulmonar con frotis positivo.21 En Lima metropolitana la tuberculosis es causa del 4,8% de las muertes. Según datos recogidos en los certificados de defunción, la tuberculosis ocupó el sexto lugar entre los casos de muerte en 1990 y el decimotercero a nivel nacional en 1997. No contamos con datos relacionados con el porcentaje total de pacientes afectados de tuberculosis del tracto digestivo aunque algunos trabajos nacionales muestran la realidad de algunos hospitales y centros de salud. ETIOPATOGENIA A pesar de las investigaciones efectuadas sobre la patogenia de la enfermedad no se tiene claro el mecanismo de la infección. Se han postulado algunos como: - La ingestión de material infectado, - Por extensión directa de órganos vecinos comprometidos. - Por diseminación hematógena o linfática. La mucosa oral intacta es extremadamente resistente a la invasión bacteriana, por lo que al localizarse el M. tuberculosis en la cavidad oral, ya sea en el esputo proveniente de una pulmonar o laríngea o en alimentos infectados, para su implantación cobra importancia la existencia de traumatismos locales. Los microorganismos de las lesiones abiertas del pulmón llegan a las vías respiratorias altas al toser y después se tragan llegando al estómago donde resisten a la acción dos y pasan al intestino delgado donde son fagocitados por el tejido linfoide, mayormente en el área ileocecal, ALUMNO: ElvisLujánRuiz
- 45. en donde se localiza el mayor porcentaje de las lesiones intestinales, a este nivel es absorbido por la mucosa intestinal y pasa hacia las placas de Peyer. El origen de la peritonitis tuberculosa es por propagación directa del intestino, por rotura de un ganglio mesentérico tuberculoso secundariamente infectado desde el intestino. La adenitis mesentérica tuberculosa es la fuente de la mayoría de las complicaciones (fístulas, peritonitis). La peritonitis tuberculosa puede también originarse por propagación de la infección de las trompas de Falopio. Algunos autores señalan que el origen hematógeno de la peritonitis tuberculosis es infrecuente. La diseminación hematógena proviene de un foco infeccioso extraintestinal distante, muchos autores lo consideran como el segundo mecanismo tanto en la tuberculosis intestinal como peritoneal. El compromiso puede darse por la diseminación hematógena a partir de un foco primario activo. Presente el bacilo tuberculoso en la pared intestinal, el c ompromiso fundamental se halla en la submucosa donde abundante tejido linfoide, la colonización estimula una respuesta inflamatoria con engrosamiento por edema, hiperplasia linfática, infiltración celular y formación de tubérculos (folículo de Koester) formado por células epiteliales, mononucleares y células gigantes o de Langhans. Con la necrosis de los tubérculos primarios, los bacilos pasan a los linfáticos intramurales y de ahí a los ganglios linfáticos regionales. A través de la vía linfática los bacilos son llevados hasta los ganglios mesentéricos, los que posteriormente presentan necrosis caseosa y calcificación. Este proceso puede dar lugar a secuelas, en algunos casos endarteritis que genera una deficiente irrigación con necrosis y ulceración de la mucosa subyacente resultando en la forma ulcerativa de la enfermedad. Al cicatrizar las úlceras éstas se fibrosan provocando estenosis del lumen y engrosamiento de la pared intestinal, finalmente una reacción fibroblástica más intensa puede darse en la submucosa y subserosa dando lugar a la forma hipertrófica del compromiso intestinal. ALUMNO: ElvisLujánRuiz
- 46. PATOLOGÍA El Mycobacterium tuberculosis puede infectar el aparato digestivo a través de la sangre, la linfa, o por contacto, sin embargo, la infección resulta principalmente de la deglución del esputo infectado. Se aprecian dos formas de presentación de la tuberculosis intestinal: - La forma ulcerosa, donde hay gran producción de tubérculos miliares que se fusionan y caseifican dando necrosis de la mucosa suprayacente, con ulceraciones a nivel de la mucosa intestinal, sin embargo, es raro la perforación de las úlceras. El M. tuberculosis desencadena una reacción inflamatoria granulomatosa en la pared intestinal con ulceración exudativa de centro caseoso necrótico, isquemia y reacción fibroblástica. Las lesiones se confinan inicialmente a los folículos linfoides del íleon y el ciego, pero más tarde rodean todo el intestino y originan linfadenopatía masiva en el mesenterio. - La forma hiperplásica crónica, que tiene preferencia por el ciego, ocasionalmente toma el íleon, y existe formación de tubérculos con caseificación, inflamación granulomatosa difusa con engrosamiento de la pared intestinal. La lesión patognomónica de la tuberculosis peritoneal es la siembra de la serosa con los tubérculos miliares, que son lesiones finas de color gris blanco. CUADRO CLÍNICO La tuberculosis de localización intestinal y/o peritoneal es un padecimiento crónico cuyos síntomas y signos son inespecíficos. La tuberculosis digestiva con sus variantes intestinal, peritoneal, enteroperitoneal, mesentérica con o sin compromiso de otros órganos puede imitar y semejar a una gran variedad de desórdenes abdominales, incluyendo patología neoplásica, infecciosa, inflamatoria inespecífica. Un evidente grado de agudeza clínica es importante para el diagnóstico. El tiempo de evolución previo a la consulta médica fluctúa entre uno y seis meses. En la tuberculosis intestinal el síntoma frecuente es el dolor abdominal, tanto espontáneo como a la palpación, es de localización difusa, a predominio del cuadrante inferior derecho y se acompaña generalmente de anorexia y náuseas. Existe alteración del patrón defecatorio con presencia de diarreas persistentes y/o alternancia de diarrea con estreñimiento, las heces pueden ser acuosas o de tipo disentérico con presencia de moco y sangre. La hematoquecia puede presentarse en la forma ulcerosa. Signos importantes son la pérdida de peso, la distensión abdominal, fiebre, y borborigmos en la fosa ilíaca derecha; también, es posible palpar masa dolorosa en el cuadrante inferior derecho (forma hiperplásica). La tuberculosis de localización peritoneal, presente mayormente en adultos jóvenes, tiene manifestaciones sistémicas más que peritoneales, con fiebre, hiporexia y malestar general. El dolor abdominal es de moderada a gran intensidad con marcada postración y ascitis, pudiendo evidenciarse el signo del tablero de ajedrez. La ascitis es el hallazgo físico más frecuente y se encuentra en forma manifiesta en el 75% de los casos. El líquido ascítico es un exudado donde el valor entre la concentración de albúmina plasmática y albúmina del líquido ascítico es menor a 1,1 g. El cociente entre el LDH del suero y el LDH del líquido ascítico es menor a 1, lo que indica que existe un mecanismo exudativo, situación que igualmente ocurre en la carcinomatosis. La elevación de la actividad de la enzima adenosina deaminasa (ADA) es de gran ayuda diagnóstica cuando los valores sobrepasan de 40 U/L. La glucosa en el líquido ascítico se encuentra disminuida en relación con la glicemia, existe celularidad incrementada, el recuento leucocitario es mayor de 500 con predominio linfocitario. El gradiente entre el pH arterial y el pH ascítico es mayor de 0, 10. ALUMNO: ElvisLujánRuiz
- 47. Es extremadamente raro encontrar un líquido ascítico hemorrágico.19Asimismo, es infrecuente el hallazgo del microorganismo en el líquido ascítico. En España, Nistal de Paz18 reportó casos de tuberculosis intestinal con compromiso ginecológico que simulaban una neoplasia ovárica, en los que se encontraron valores de marcador tumoral para carcinoma de ovario, Ca 125, elevados, dentro de los rangos para malignidad. La elevación de los valores de Ca 125 debe inducir al diagnóstico de neoplasia maligna de ovario; sin embargo, se considera obligatorio la búsqueda de microorganismos (del Mycobacterium y de Actinomyces sp.) y la obtención de tejido para biopsia con la finalidad de hallar granulomas. Esta situación también ha sido mencionada en otros estudios.18 Cuando existe disminución de peso, dolor abdominal y diarrea en pacientes con tuberculosis pulmonar se debe sospecharse la existencia de compromiso enteroperitoneal. En una revisión de 335 historias clínicas, de las cuales 140 cumplieron con los criterios de inclusión para el estudio, efectuado entre los años 1969 y 1987 en el Hospital Nacional Cayetano Heredia, en pacientes mayores de 14 años diagnosticados de tuberculosis gastrointestinal y/o peritoneal y tuberculosis generalizada, el estudio conc luyó que el 48,5% tuvo evidencia de compromiso gastrointestinal, 18,5% presentó evidencia de compromiso tanto gastrointestinal como peritoneal y 33% fue catalogado como de localización peritoneal únicamente. La mayoría de casos fue en pacientes menores de 40 años (63% del total de pacientes). La tuberculosis intestinal y peritoneal se asoció a la localización pulmonar en el 84% de los casos y a mortalidad en el 14,5% de los pacientes. En 1986, Chahud10 reportó 30,1 % de tuberculosis peritoneal y 10,8% de tuberculosis intestinal más peritoneal. Un estudio retrospectivo de cinco años (1993-1998) efectuado en el HNERM (EsSalud), en un total de 58 historias clínicas, reportó cifras más altas de compromiso peritoneal, sea aislado o peritoneal más intestinal, en cuyo diagnóstico ha jugado un papel preponderante la laparoscopia diagnóstica. Este estudio consideró que el diagnóstico de la tuberculosis digestiva sigue siendo tedioso, largo y costoso y que se requiere de exámenes invasivos para el diagnóstico definit ivo en la totalidad de casos revisados. Señaló que durante los años ochenta y mediados de los noventa existía una ligera tendencia a la baja en la incidencia de tuberculosis pulmonar en el Perú; sin embargo, las localizaciones extrapulmonares cobraron cada vez mayor importancia. Celestino8 mencionó que más del 80% de casos de las series de pacientes con tuberculosis gastrointestinal estudiada tuvo enfermedad pulmonar activa, y dio relevancia a los métodos radiológicos contrastados de intestino delgado y de colon para el diagnóstico de esta enfermedad. Señaló que cuando la localización es en colon, íleon distal o ileocecal, la colonoscopia con biopsia, y con "impronta" para la coloración en lámina, fue de alto rendimie nto para encontrar el bacilo y el granuloma caseoso. En casos dudosos se puede, aseveró, emplear la laparotomía exploratoria o la prueba terapéutica. ALUMNO: ElvisLujánRuiz
- 48. REFERENCIAS BIBLIOGRÁFICAS .STEPHEN MCPHEE. FISIOPATOLOGIA DE LA ENFERMEDAD. EDITORIAL MCGRAWHILL. 6 EDICION. PAG.373 .HARRINSON. MEDICINA INTERNA. EDITORIAL MCGRAWHILL, 9 EDICION. PAG 560 GONZÁLEZ GARCÍA M.A., GARCÍA SÁNCHEZ M., ALVAREZ MÁRQUEZ A. PRINCIPIOS DE URGENCIAS, EMERGENCIAS Y CUIDADOS CRÍTICOS. CAPÍTULO 3. 2. HEMORRAGIAS DIGESTIVAS UNINET DRS. GERMÁN CABRERA ROMERO, VÍCTOR MACEDO PEÑA. HEMORRAGIA DIGESTIVA. TOMO 1 CAPITULO 10 JAWETZ, MELNICK Y ALBERG. “MICROBIOLOGÍA MEDICA”. 26° ED. MEXICO. MCGRAW- o HILL INTERAMERICANA EDITORES.CAP. 35. PAG.511-526 PATRICK R. MURRAY, KEN S. ROSENTHAL, MICHAEL A. PFALLER “MICROBIOLOGÍA MEDICA”.7° ED. ESPAÑA.ELSEVIER.2013. CAP.63. PAG:592-594 HAMMER G. MCPHEES. FISIOPATOLOGÍADE LA ENFERMEDAD.7 ED. MEXICO:MC GRAW HILL; 2015. GROSSMAN S. MATTSON C. FISIOPATOLOGIA PORTH.9 EDICION. BARCELONA: WOLTERS KLUWER; 2014 MONTOYA ANA.RESTEPRO JUAN. HEPATITIS C (LA CLINICA Y EL LABORATORIO). MEDICINA Y LABORATORIO.2011. VOLUMEN17.NUMEROS 9-10. (SEDE WEB) DISPONIBLE EN: HTTP://WWW.MEDIGRAPHIC.COM/PDFS/MEDLAB/MYL-2011/MYL119-10B.PDF CASANOVA RITUERTO. CASANOVAS TERESA. HEPATITIS POR EL VIRUS DE LA HEPATITIS C. SERVICIOS DE MICROBIOLOGÍA Y GASTROENTEROLOGÍA, CIUDAD SANITARIA Y UNIVERSITARIA DE BELLVITGE. HOSPITALET DE LLOBREGAT (BARCELONA) (SEDE WEB) DISPONIBLE EN: HTTPS://WWW.SEIMC.ORG/CONTENIDOS/CCS/REVISIONESTEMATICAS/SEROLOGIA/VH C.PDF R.GARCIA CALVO, LITIASIS BILIAR COMPLICADA, REVISTA DE LA SOCIEDAD VALENCIANA DE PATOLOGÍA DIGESTIVA, 2001, 4 PAGINAS, DISPONIBLE EN: HTTP://WWW.ELSEVIER.ES/ES-REVISTA-REVISTA-SOCIEDAD-VALENCIANA- PATOLOGIA-DIGESTIVA-160-ARTICULO-COLEDOCOLITIASIS-13032137 CONSUELO QUINTANILLA L, HUMBERTO FLISFISH F. COLEDOCOLITIASIS. REVISTA DE MEDICINA Y HUMANIDADES. VOLUMEN 1. 2009. 9 PAGINAS MERCAKMANUALS.COM. ESTADOS UNIDOS, ELDON A.SHAFFER, 18 DE SETIEMBRE DE 2016, DISPONIBLE EN: HTTP://WWW.MERCKMANUALS.COM/ES- US/PROFESSIONAL/TRASTORNOS-HEP%C3%A1TICOS-Y-BILIARES/TRASTORNOS- DE-LA-VES%C3%ADCULA-BILIAR-Y-LOS-CONDUCTOS- BILIARES/COLEDOCOLITIASIS-Y-COLANGITIS UPTC-UNAL.BLOGSPOT.PE. BOGOTA-COLOMBIA, ALEX CASALLAS, MEDICINA INTERNA- COLEDOCOLITIAIS, 27 DE AGOSTO DE 2012; 18 DE SETIEMBRE DE 2016,
- 49. DISPONIBLE EN: HTTP://UPTC- UNAL.BLOGSPOT.PE/2012/08/COLEDOCOLITIASIS.HTML RIVERÓN CORTEGUERA, R. L. (1999). FISIOPATOLOGÍA DE LA DIARREA AGUDA. REVISTA CUBANA DE PEDIATRÍA, 71(2), 86-115. FERNANDEZ, S., & ALONSO, G. (2009). COLERA Y VIBRIO CHOLERAE. REV. INST. NAC. HIG, 40(2), 50-69. ROBLES, L. A., GARCÍA, R. M., & LÓPEZ, J. T. (1999). TOXINAS DE VIBRIO CHOLERAE. UNA REVISIÓN. REVISTA LATINOAMERICANA DE PATOLOGÍA CLÍNICA Y MEDICINA DE LABORATORIO, 46(4), 255-259. VÁZQUEZ, E. G., TORRES, A. H., MARTÍNEZ, J. H., & GÓMEZ, J. G. (2014). CÓLERA Y OTRAS INFECCIONES DEL GÉNERO VIBRIO. MEDICINE-PROGRAMA DE FORMACIÓN MÉDICA CONTINUADA ACREDITADO, 11(56), 3317-3321. ARENAS, C., DOBLAS, A., JURADO, R., RIVERO, A., TORRE, J. (2010).FIEBRE TIFOIDEA Y OTRAS INFECCIONES POR SALMONELLAS. MEDICINE, 10(52):3497-501. (2016). AIBARRA.ORG. REVISADO EL 14 DE SEPTIEMBRE DE 2016, DESDE HTTP://WWW.AIBARRA.ORG/APUNTES/CRITICOS/GUIAS/INFECCIOSOS/SALMON ELLOSIS.PDF GAVILÁN, C., GARCÍA, B., GONZALES, R. (2011) GASTROENTERITIS AGUDA CAPITULO 13. PROTOCOLOS DIAGNÓSTICO-TERAPÉUTICOS DE LA AEP: INFECTOLOGÍA PEDIÁTRICA. ANTHONY S. FAUCI, EUGENE BRAUNWALD, DENNIS L. KASPER. PRINCIPIOS DE MEDICINA INTERNA.VOL 3 19ª ED. MÉXICO DF: MC-GRAW HILL INTERAMERICANA EDITORES; 2016, PÁG. 852-856. ESCHERICHIA COLI ENTEROPATÓGENA (EPEC): UNA CAUSA FRECUENTE DE DIARREA INFANTIL. (2003). SALUD EN TABASCO. [ONLINE] AVAILABLE AT: HTTP://WWW.REDALYC.ORG/ARTICULO.OA?ID=48709108 [ACCESSED 15 SEP. 2016]. DRA. RAQUEL FERNÁNDEZ FERRÁN, DR. CARLOS RODRÍGUEZ PÉREZ,DRA. ISIS DE LOS A. RODRÍGUEZ RIBALTA Y DR. FREDDY GÓMEZ MARTÍNEZ; BVS.SLD.CU. ESCHERICHIA COLI COMO CAUSA DE DIARREA INFANTIL. (2010). [ONLINE] AVAILABLE AT: HTTP://BVS.SLD.CU/REVISTAS/PED/VOL75_3_03/PED10303.HTM [ACCESSED 13 SEP. 2016]. TRATADO.UNINET.EDU. 3.3.5. ETIOLOGIA. [ONLINE] AVAILABLE AT: HTTP://TRATADO.UNINET.EDU/C030305.HTML (2014). [ACCESSED 14 SEP. 2016]. FERNÁNDEZ FERRÁN, R., RODRÍGUEZ PÉREZ, C., RODRÍGUEZ RIBALTA, I. AND GÓMEZ MARTÍNEZ, F. ESCHERICHIA COLI COMO CAUSA DE DIARREA INFANTIL. REVISTA CUBANA DE PEDIATRÍA, (2003). [ONLINE] 75(3), PP.0-0. AVAILABLE AT: HTTP://SCIELO.SLD.CU/SCIELO.PHP?SCRIPT=SCI_ARTTEXT&PID=S0034- 75312003000300010 [ACCESSED 14 SEP. 2016]. BAGGINI, S. AND BAGGINI, S. LAS ENTEROBACTERIAS (PARTE 2). [ONLINE] BAGGINIS.BLOGSPOT.PE. (2014). AVAILABLE AT: HTTP://BAGGINIS.BLOGSPOT.PE/2014/08/LAS-ENTEROBACTERIAS-PARTE-2.HTML [ACCESSED 16 SEP. 2016].
- 50. VERSIÓN EN INGLÉS REVISADA POR: SUBODH K. LAL, A. ENTERITIS POR E. COLI: MEDLINEPLUS ENCICLOPEDIA MÉDICA. [ONLINE] MEDLINEPLUS.GOV. (2016). AVAILABLE AT: HTTPS://MEDLINEPLUS.GOV/SPANISH/ENCY/ARTICLE/000296.HTM [ACCESSED 15 SEP. 2016]. THERESA J. OCHOA WOODELL , DIARREA PRODUCIDA POR ESCHERICHIA COLI. (2014). [ONLINE] AVAILABLE AT: HTTP://SISBIB.UNMSM.EDU.PE/BVREVISTAS/SPEIT/2006_N2/PDF/A03.PDF [ACCESSED 13 SEP. 2016]. WILLIAM F. GANONG. FISIOPATOLOGÍA MÉDICA. EDITORIAL MANUAL MODERNO. 5ª EDICIÓN. PAG 421 HTTP://SISBIB.UNMSM.EDU.PE/BVREVISTAS/SPMI/V15N1/TUBER_INTEST_PERIT O.HTM HTTP://SISBIB.UNMSM.EDU.PE/BVREVISTAS/GASTRO/VOL_17S1/TUBERC_GASTRO INT.HTM