Descargado 190 veces































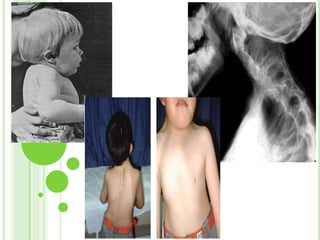







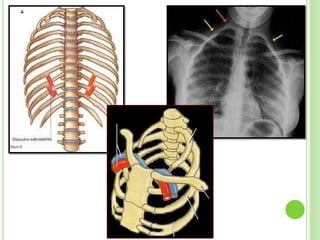






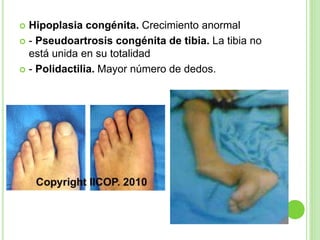



Este documento resume diferentes tipos de malformaciones óseas, incluyendo anomalías óseas generalizadas como el gigantismo, osteopetrosis y osteogénesis imperfecta; malformaciones craneanales como la craneosinostosis, microcefalia y síndrome de Crouzon; malformaciones de la columna vertebral como la espina bífida y la espondilolistesis; y malformaciones del tórax como el tórax excavado y en quilla. El documento describe las características clínicas de cada condición.























































































