Descargado 78 veces

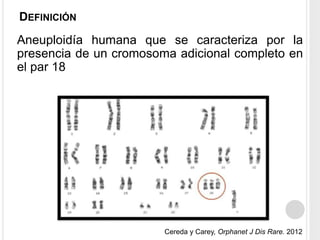



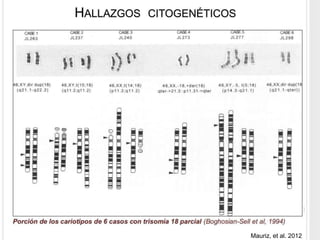


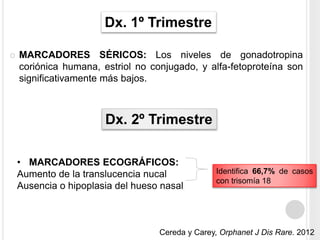
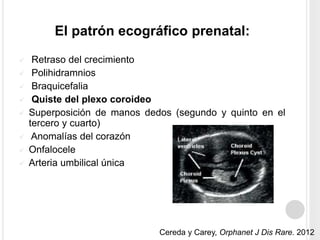
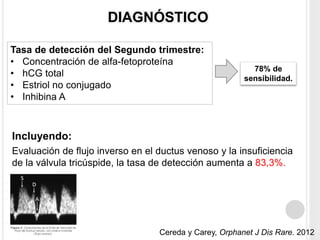

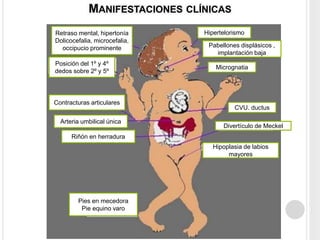
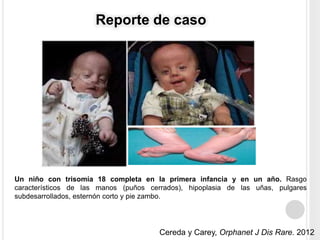
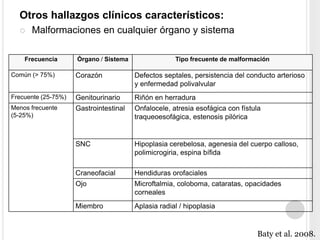

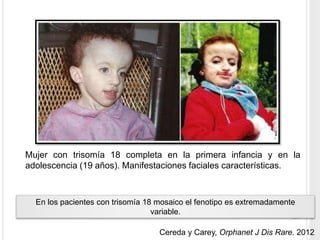



La trisomía 18 o síndrome de Edwards es una aneuploidía causada por la presencia de un cromosoma 18 adicional. Se caracteriza por defectos en el crecimiento y malformaciones en múltiples órganos. El diagnóstico prenatal incluye análisis de marcadores séricos y hallazgos ecográficos característicos. La supervivencia media es baja, entre 3 a 14,5 días, aunque algunos pacientes pueden vivir más de un año.




















































































