Descargado 217 veces










































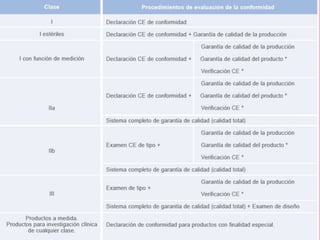








Este documento proporciona información sobre productos sanitarios no farmacéuticos. Define qué son los productos sanitarios y accesorios, y explica su clasificación de riesgo. También cubre la legislación aplicable, requisitos esenciales como la seguridad y esterilización, y conceptos como tarjetas de implantación. El documento proporciona detalles sobre la regulación de estos productos en España.























































































