
![Estados protromboticos
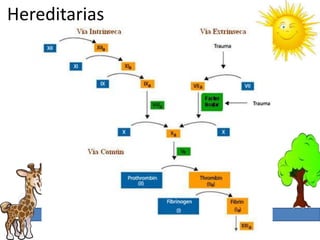
Congénitos
Deficiencia ATIII, Prot. C,S, Plasminogeno.
Resistencia a factor V de Leyden.
[] pro coagulante factor VIII gen protrombina
Lesión endotelial homosisteina](https://image.slidesharecdn.com/trastornosplaquetarios-120929162932-phpapp01/85/Trastornos-plaquetarios-3-320.jpg)



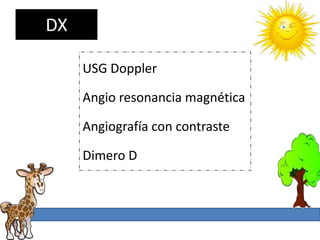

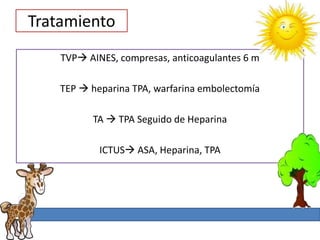
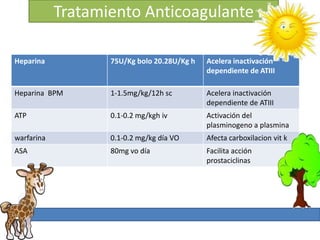




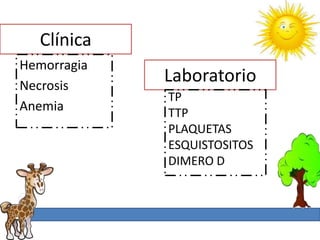
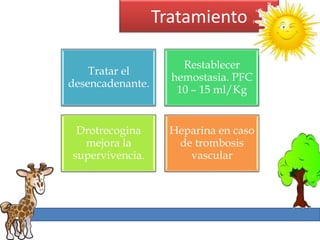


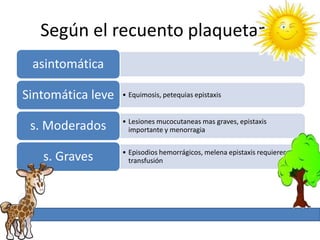


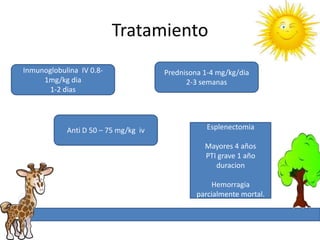

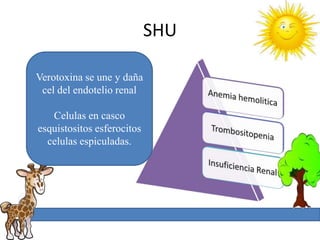
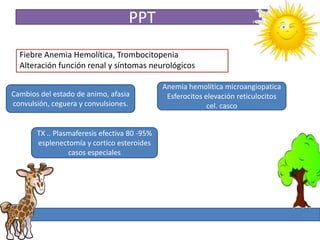
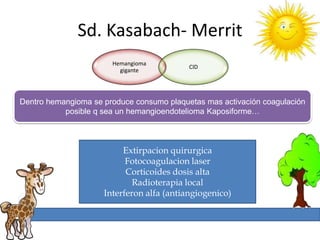

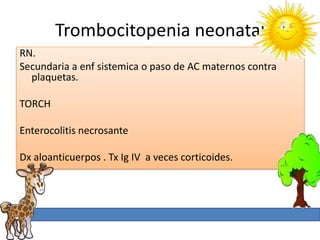
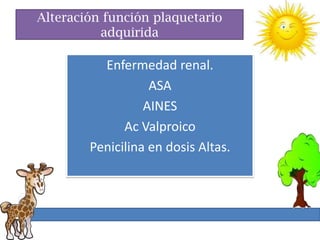

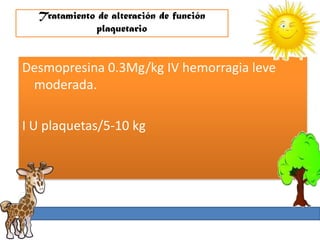
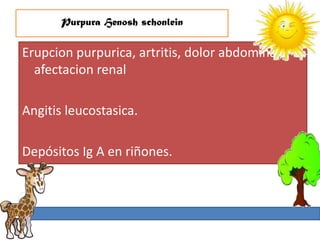
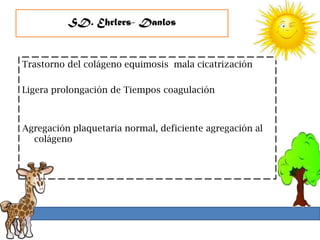


1. La trombosis puede ser hereditaria o adquirida, y puede ocurrir debido a estados protrombóticos congénitos o adquiridos. 2. Los estados protrombóticos congénitos incluyen deficiencias de proteínas anticoagulantes y resistencia al factor V de Leyden, mientras que los estados protrombóticos adquiridos incluyen inmovilización, cirugías, embarazos y síndrome nefrótico. 3. El diagnóstico incluye ecografía Doppler, reson
























![[20] hemoderivados](https://cdn.slidesharecdn.com/ss_thumbnails/20hemoderivados-130703203116-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)
























































