Descargado 314 veces



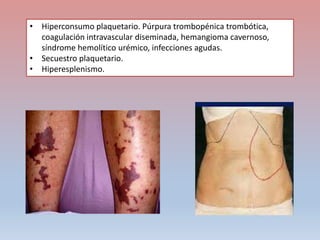

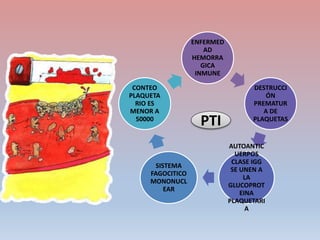

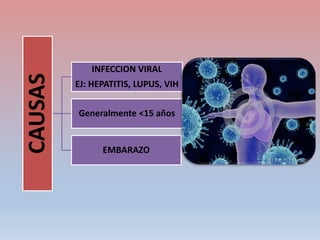
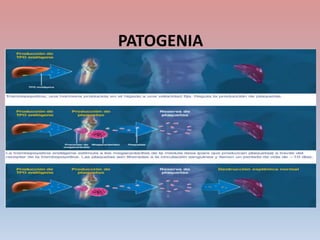
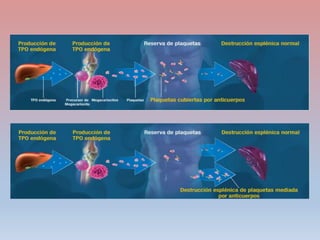
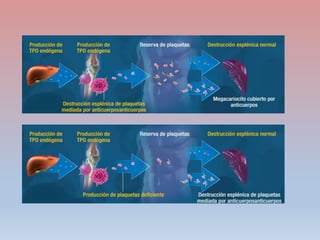
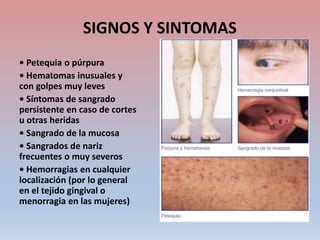






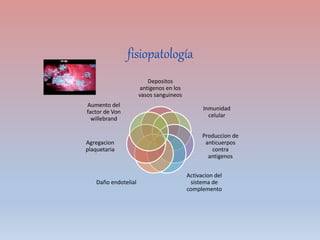



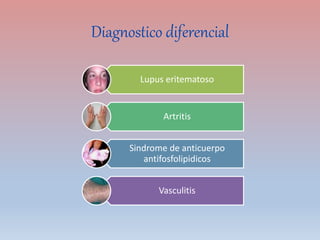



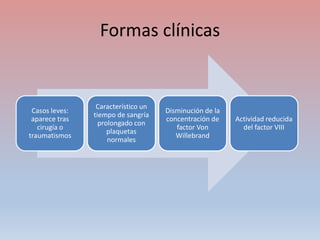


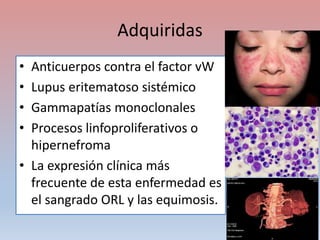



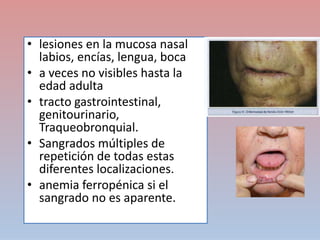


Este documento describe diferentes tipos de trombopenia, incluyendo sus causas, síntomas y tratamientos. La trombopenia se define como un recuento de plaquetas por debajo de 100,000 plaquetas/mm3 y puede deberse a una hipoproducción o disminución de la supervivencia de las plaquetas. Las causas incluyen enfermedades de la médula ósea, fármacos, infecciones y trastornos autoinmunes. El tratamiento depende de la causa subyacente.







































































































