Descargado 216 veces










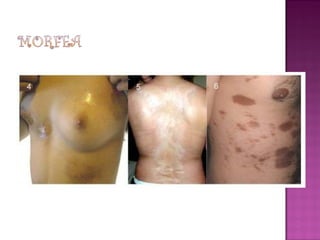
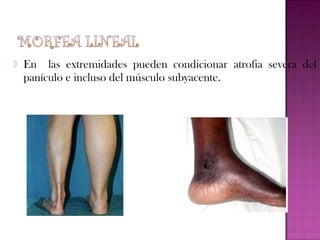
















La esclerodermia es una enfermedad autoinmune del tejido conectivo que se caracteriza por fibrosis e induración progresiva de la piel y posible afectación de órganos internos. Puede presentarse como morfea localizada o como esclerosis sistémica. Su tratamiento suele ser difícil y los resultados modestos, aunque los bloqueadores de canales de calcio mejoran el fenómeno de Raynaud y la fototerapia puede usarse en formas extensas.
































































![Esclerodermia[1].pptx lesiones tejido dermis y epidermis, reumatologia](https://cdn.slidesharecdn.com/ss_thumbnails/esclerodermia1-250401013807-d8d624f6-thumbnail.jpg?width=640&height=640&fit=bounds)






















