Descargado 166 veces

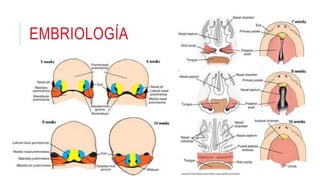
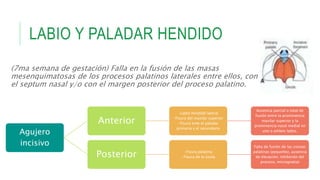
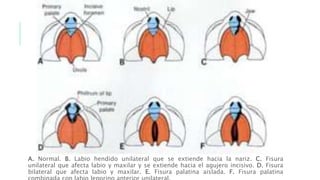

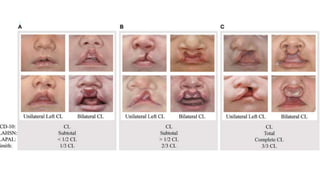
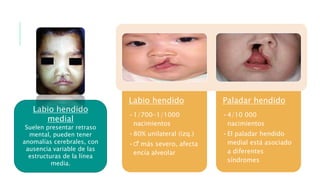

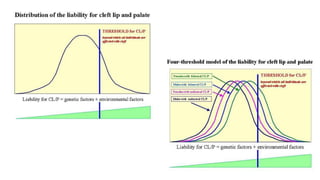
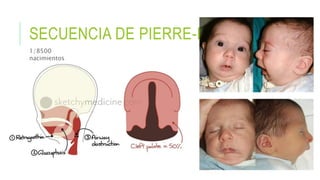

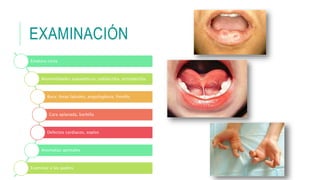
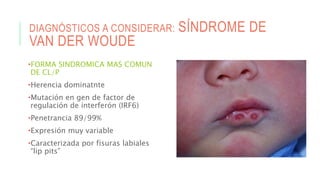

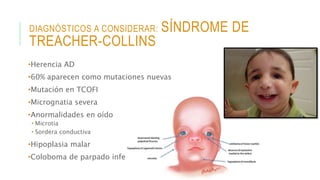
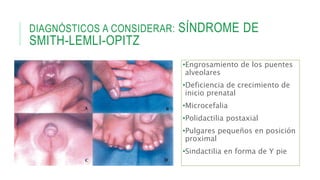
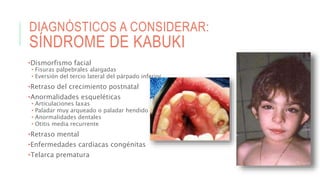
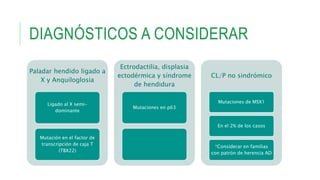
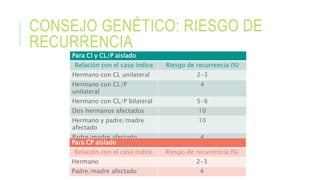
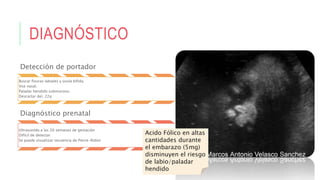
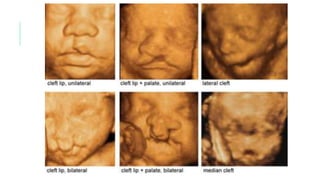



Este documento describe la embriología, características clínicas, diagnósticos diferenciales y consejo genético relacionados con el labio y paladar hendido. Explica que estos defectos ocurren debido a fallas en la fusión de las estructuras faciales durante la 7ma semana de gestación. Provee detalles sobre varios síndromes asociados como Van der Woude, Stickler y Treacher-Collins. Recomienda exámenes genéticos para determinar el riesgo de recurrencia, especialmente si hay antecedentes


































































































