Descargado 10 veces



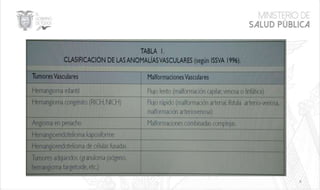

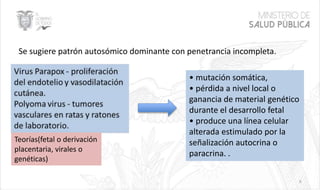
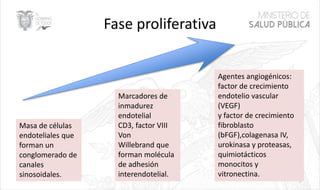
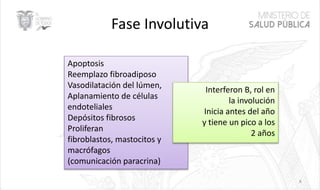
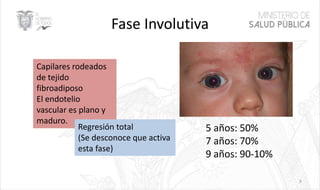





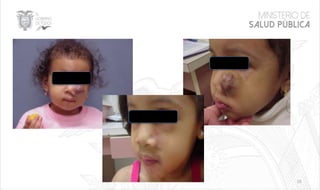



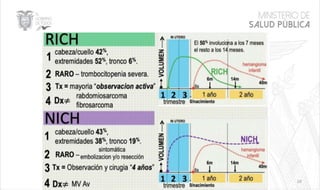

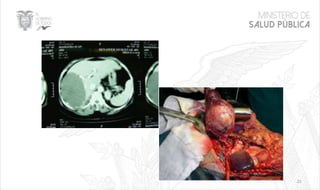

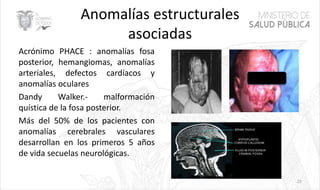







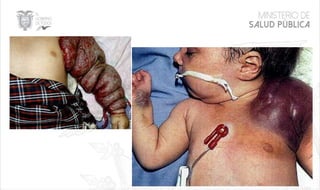
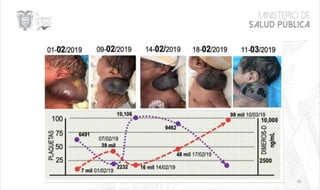
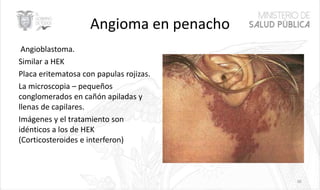
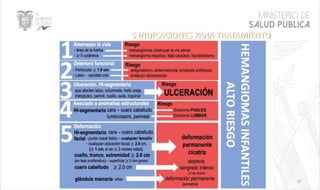



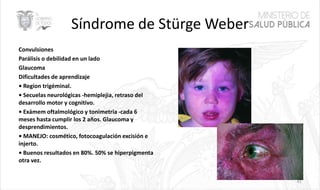
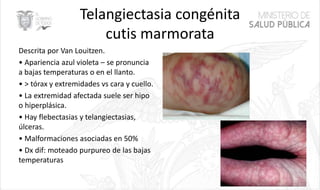


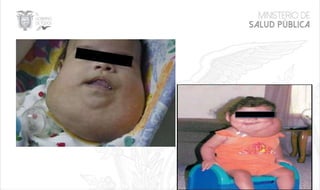

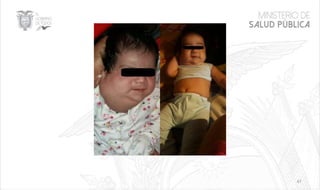





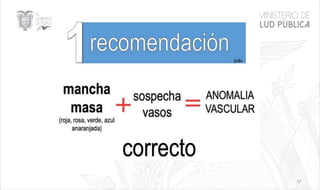
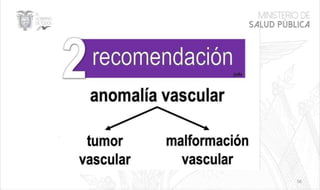




Este documento resume las principales características de las malformaciones vasculares en pediatría. Describe el hemangioma infantil, su patogénesis, fases de crecimiento y regresión, manifestaciones clínicas y tratamientos. También cubre otras malformaciones como las capilares, linfáticas y síndromes asociados como Stürge Weber. El documento ofrece una guía para el diagnóstico y manejo de estas afecciones en niños.














































![Malformaciones arteriovenosas[1]](https://cdn.slidesharecdn.com/ss_thumbnails/malformacionesarteriovenosas1-100618180014-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)







































