Descargado 14 veces


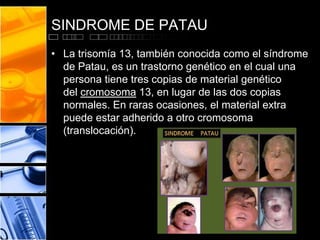



















Este documento presenta información sobre varios síndromes cromosómicos, incluidos los síndromes de Patau, Cri-du-chat, Down, Gilbert y Turner. Describe las causas, síntomas y características de cada uno, así como posibles pruebas de diagnóstico y formas de prevención cuando sea posible. El documento proporciona detalles sobre las mutaciones cromosómicas y cómo afectan el desarrollo físico y mental.



































































