Descargado 33 veces




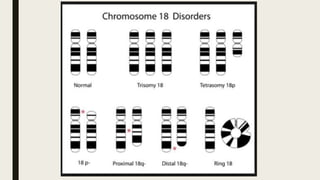

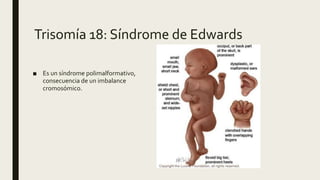
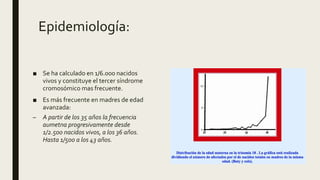



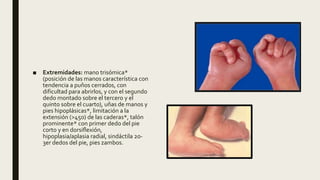


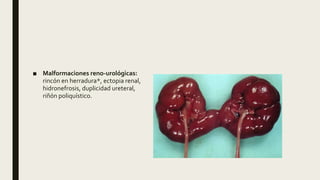
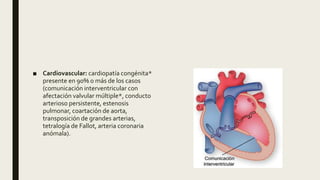








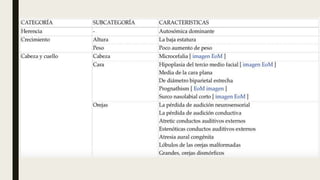


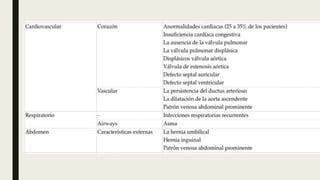
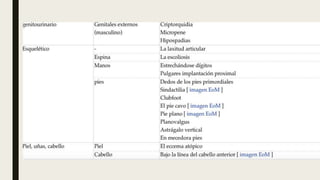
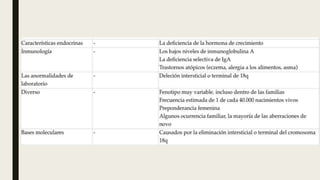

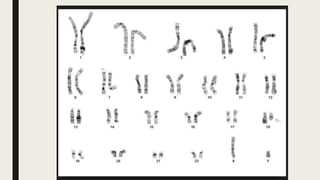
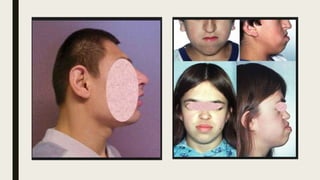


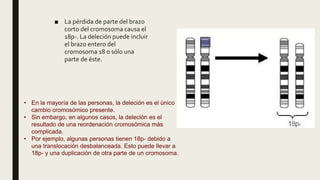
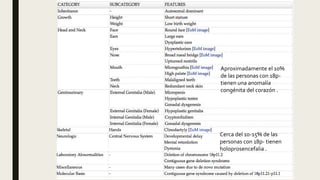


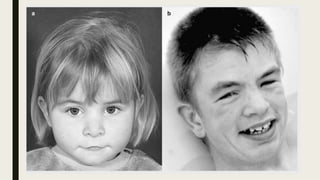





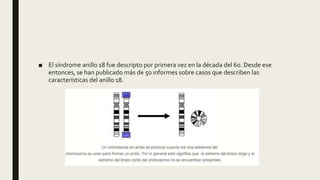





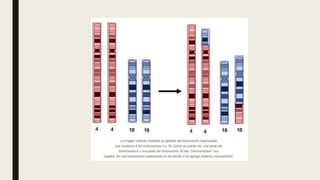
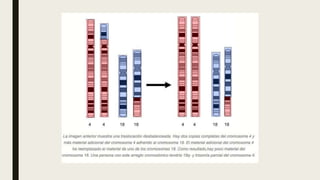
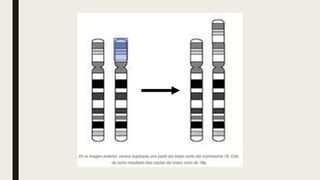



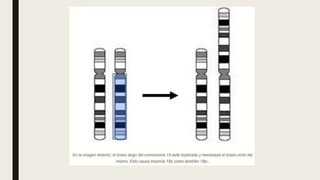

El documento describe diferentes anomalías cromosómicas relacionadas con el cromosoma 18. Se detalla información sobre la trisomía 18 o síndrome de Edwards, la deleción 18q, la deleción 18p, el síndrome de Pitt-Hopkins y la tetrasomía 18p, entre otras. Todas estas anomalías se asocian con características fenotípicas particulares y retraso intelectual.



































































