Descargado 10 veces




























![MECANISMOS DE ACCIÓN
Los mecanismos precisos responsables de la capacidad de los agentes inhalados volátiles potentes (por
ejemplo, el sevoflurano , desflurano , y el isoflurano) para inducir la anestesia no se conocen.
Canales iónicos en el sistema nervioso central se ven afectados por los agentes volátiles
→ ácido gamma-aminobutírico A [GABA A ], glicina, y los receptores de glutamato
N2O → antagonismo de la N-metil -D-aspartato (NMDA).](https://image.slidesharecdn.com/opiodessubir-161019113046/85/Opiodes-31-320.jpg)










![ISOFLURANO
El isoflurano es un agente anestésico general capaz de producir una profunda
depresión respiratoria.
Alteran la actividad de los canales neuronales de iones, en particular los receptores
de neurotransmisores sinápticos rápidos (acetilcolina nicotínico, ácido gamma-
aminobutírico [GABA] y receptores de glutamato).
Puede deprimir la contractilidad miocárdica → disminución de la presión arterial a
través de una disminución de la resistencia vascular sistémica, y disminuir la
actividad nerviosa simpática.
Mecanismo de acción](https://image.slidesharecdn.com/opiodessubir-161019113046/85/Opiodes-42-320.jpg)



![BIBLIOGRAFÍA
Fletcher D. Farmacología de los opioides. EMC - Anest-Reanim [Internet]. 2011 [citado 5 de
septiembre de 2016];37(2):1-24. Disponible en:
http://www.sciencedirect.com/science/article/pii/S1280470311710313.
Chabner B, Brunton L, Knollman B. Goodman and Gilman’s The Pharmacological Basis of
Therapeutics, Twelfth Edition. McGraw-Hill Education; 2011. 1808 p.
Anestesia P por SJ de D. Investigación Anestesia: Receptores opioides [Internet]. [citado 5 de
septiembre de 2016]. Disponible en: http://investigacionanestesia.blogspot.com/2009/10/en-
1973-tres-grupos-de-investigacion.html](https://image.slidesharecdn.com/opiodessubir-161019113046/85/Opiodes-46-320.jpg)


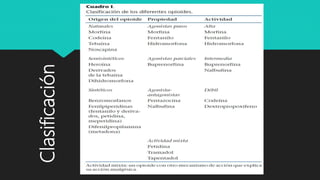
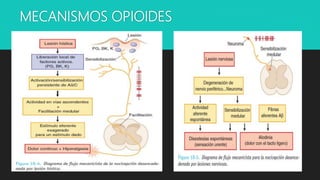
Este documento presenta información sobre los opioides, incluyendo su historia, sistemas endógenos, receptores, clasificación, mecanismos de acción y efectos farmacológicos. Describe los principales opioides exógenos como la morfina, fentanilo, tramadol y codeína, y su farmacocinética y uso clínico. También cubre brevemente los anestésicos inhalatorios y su mecanismo de acción.









































































