













![Fuente Bibliográfica
Ramachandran, Manoj. Osteogénesis Imperfecta. Medscape Reference [en
línea]. Junio 2012. Disponible en:
http://emedicine.medscape.com/article/1256726
New Perspectives on Osteogenesis Imperfecta por Antonella Forlino [et al].
Medscape Education [en línea]. Junio 2011. Disponible en:
http://www.medscape.org/viewarticle/744153
Understanding the Structure of Bones. Osteogenesis Imperfecta
Foundation [en línea]. Disponible en:
http://www.oif.org/site/PageServer?pagename=BoneStruct
How do health care providers diagnose osteogenesis imperfecta?.
NICHD [en línea]. Noviembre 2011. Disponible en:
http://www.nichd.nih.gov/health/topics/organogenesisimp/conditioni
nfo/pages/diagnose.aspx](https://image.slidesharecdn.com/osteognesisimperfectaupmed-130629171008-phpapp01/85/Osteogenesis-Imperfecta-UP-Med-24-320.jpg)

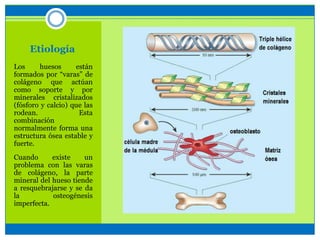
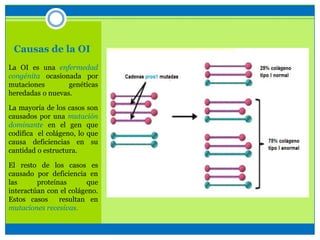
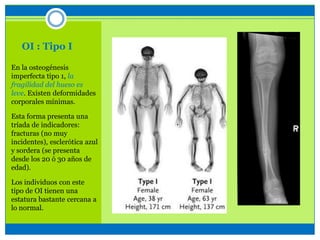
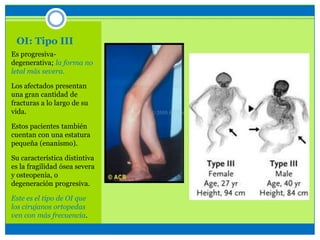
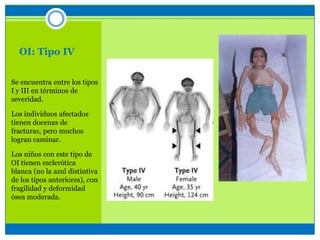
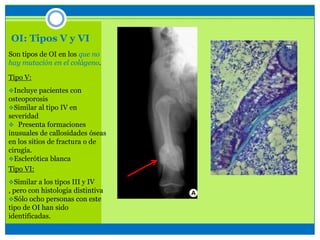
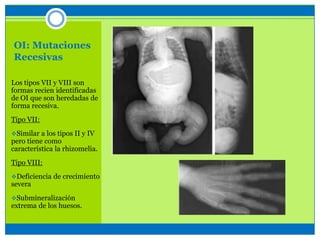
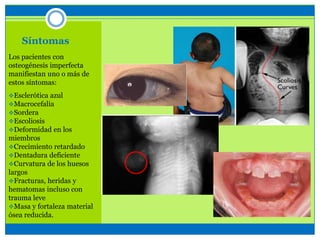
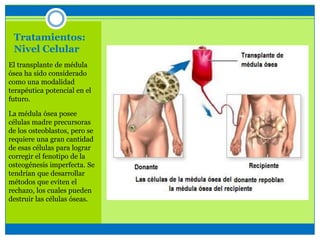
La osteogénesis imperfecta es un desorden genético del tejido conectivo óseo causado por defectos en los genes que codifican el colágeno, lo que resulta en huesos débiles y fracturables. Existen varios tipos dependiendo de la gravedad, desde leve con pocas fracturas hasta mortal al nacer. El diagnóstico se basa en exámenes físicos, radiografías y análisis genéticos. El tratamiento incluye cirugía, fisioterapia, bifosfonatos y suplementos para for























































































