Descargado 65 veces












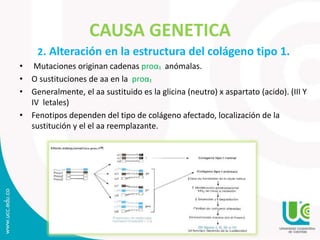












Este documento presenta tres casos clínicos de pacientes con osteogénesis imperfecta (OI). La OI es una enfermedad genética causada por mutaciones en los genes COL1A1 y COL1A2 que codifican el colágeno tipo I y producen fragilidad ósea. Los pacientes presentan múltiples fracturas desde temprana edad, baja estatura y deformidades óseas. El documento también describe los síntomas, diagnóstico, causas genéticas y tratamiento de la OI.























































































