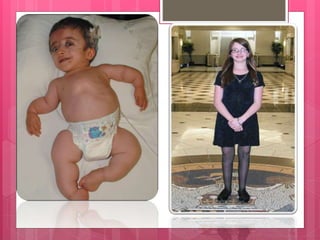
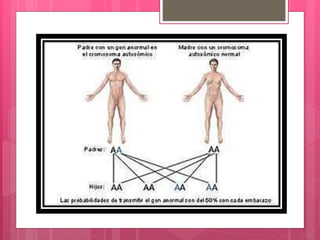
La osteogénesis imperfecta, o enfermedad de los huesos de cristal, es un trastorno congénito que provoca una fragilidad ósea extrema debido a mutaciones en los genes que codifican el colágeno. Se clasifica en varios tipos, cada uno con características diferentes, y el diagnóstico puede realizarse mediante estudios radiológicos y genéticos. No existe cura, pero hay tratamientos que alivian los síntomas y ayudan a mantener la calidad de vida de los pacientes.


















































































![Enfermedades oseas generalidades.dr elvisac [modo de compatibilidad]](https://cdn.slidesharecdn.com/ss_thumbnails/enfermedadesoseasgeneralidades-drelvisacmododecompatibilidad-110428205237-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)

























