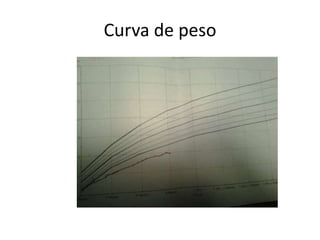
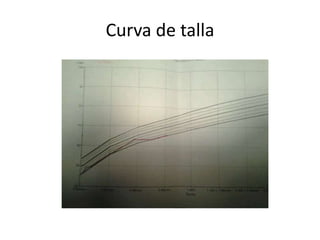
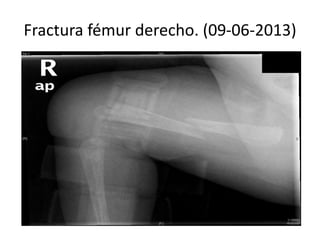
Este documento presenta el caso de una niña de 8 meses diagnosticada con osteogénesis imperfecta. Tuvo múltiples fracturas espontáneas desde los 1,5 meses de edad. Los estudios genéticos mostraron una mutación en el gen COL1A1. Se describe su tratamiento multidisciplinario incluyendo bifosfonatos, fisioterapia y cirugía ortopédica. La osteogénesis imperfecta es una enfermedad hereditaria del tejido conectivo que causa fragilidad ósea extrema y fracturas frecu
























![Clasificación de Sillence
Sillence, D. O. & Rimoin, D. L. Classification of osteogenesis imperfecta. Lancet 1978;1:
1041-1042.[Pubmed]
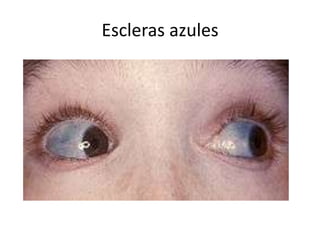
• Tipo I, la forma más leve y más frecuente.
Deformidades óseas son ligeras o nulas y la estatura
es normal. Las escleróticas son azules y hay fracturas
múltiples. Sordera frecuente manifestada a partir de
la segunda década de la vida.
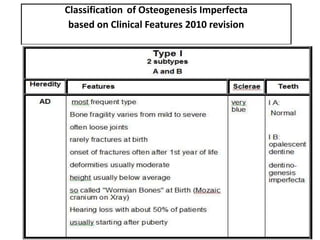
• Tipo II, letal en el período perinatal, es el de mayor
gravedad. Escasa mineralización evidenciando
fracturas intraútero y durante el parto.](https://image.slidesharecdn.com/osteogenesisimperfecta-140227062543-phpapp01/85/Osteogenesis-imperfecta-30-320.jpg)



































































![Enfermedades oseas generalidades.dr elvisac [modo de compatibilidad]](https://cdn.slidesharecdn.com/ss_thumbnails/enfermedadesoseasgeneralidades-drelvisacmododecompatibilidad-110428205237-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)





















































