Descargado 40 veces








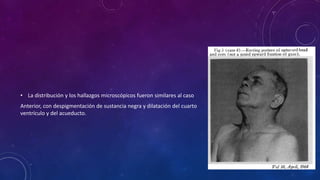







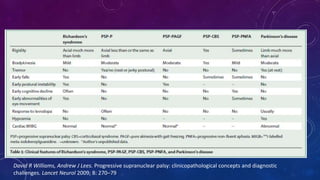

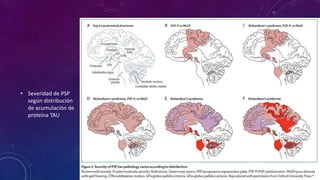









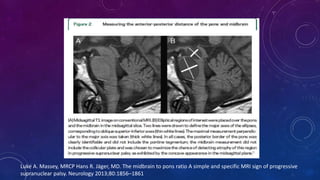
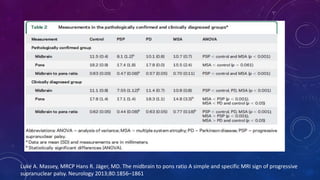


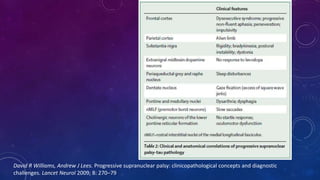
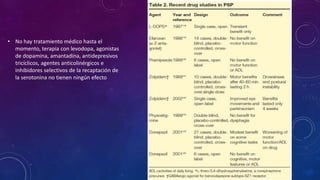


El documento describe el primer reporte de la parálisis supranuclear progresiva (PSP) realizado en 1964. Los autores describieron los síntomas cardinales de la enfermedad incluyendo parálisis de la mirada vertical, parálisis pseudobulbar y distonia axial, así como hallazgos neuropatológicos de pérdida neuronal y acumulación anormal de proteína tau. La PSP se caracteriza por ser una enfermedad neurodegenerativa progresiva que comienza después de los 40 años y causa parkinsonismo, trastornos o






























































