Descargado 62 veces







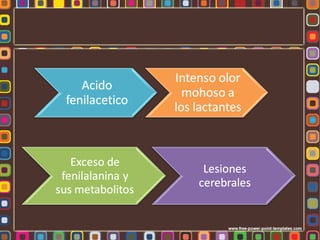


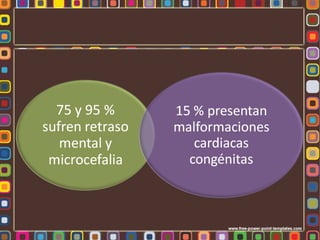



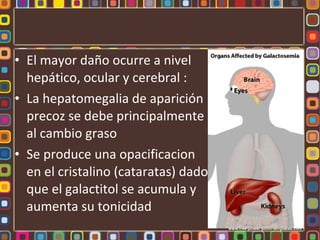
















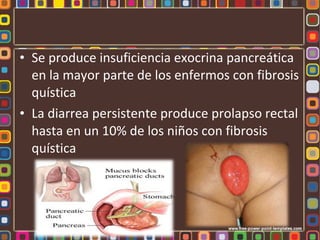












La fenilcetonuria (FCN) y la galactosemia son errores congénitos del metabolismo causados por mutaciones genéticas que alteran las enzimas fenilalanina hidroxilasa y galactosa-1-fosfato uridiltransferasa, respectivamente. Estos defectos metabólicos, de herencia autosómica recesiva, pueden causar retraso mental si no se controlan mediante dieta. La fibrosis quística se debe a mutaciones en el gen CFTR que codifica un canal de cloro, lo que altera la secreción de moco e induce in











































































