

![FISIOPATOLOGÍA
< [sales biliares] en intestino proximal
•Malabsorción grasa, calcio y vitaminas
liposulubles
Retención de sustancias normalmente
eliminadas por bilis
•Bilirrubina
•Ácidos biliares
•Colesterol
Daño hepático progresivo
•Cirrosis biliar
•Hipertensión portal
•Fallo hepático
Hondal N, Silverio C, Núñez A, Ayllón L. Colestasis en el recién nacido. Orientación diagnóstica. An Pediatr2010;58(2):162-67.](https://image.slidesharecdn.com/colestasisneonatal-170605000520/85/Colestasis-neonatal-3-320.jpg)





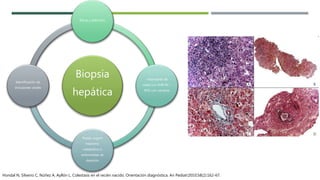




La colestasis neonatal es una condición en la que hay una alteración en el flujo biliar que causa elevación de bilirrubina y ácidos biliares en la sangre. Puede deberse a inmadurez hepática, afecciones neonatales graves, infecciones, obstrucción biliar u otras causas intrínsecas. Los síntomas incluyen ictericia, acolia, hipocolia y coluria. El diagnóstico se basa en análisis de sangre, ultrasonido hepático y otros exámenes. El tratamiento puede ser específico























































































