Descargado 301 veces


















![Síndrome de Rett
(RTT;[OMIM 312750])
Grave enfermedad del neurodesarrollo, que afecta principalmente a
niñas.
Constituye la segunda causa de retraso mental profundo más
frecuente en mujeres después del síndrome de Down, con una
incidencia de 1: 10.000 de recién nacidas.
En España se considera que hay 2500 niñas afectadas y en
Cataluña 350.](https://image.slidesharecdn.com/sndromederett-140915203602-phpapp01/85/Sindrome-de-Rett-21-320.jpg)







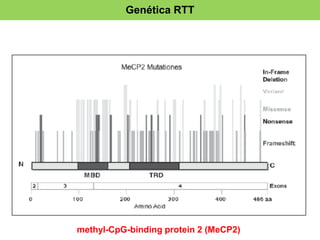


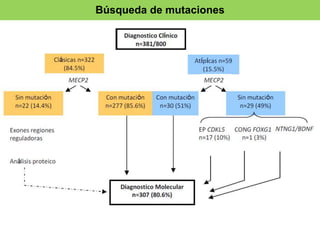




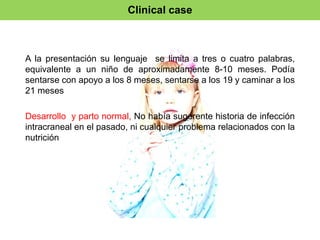
El documento presenta un caso clínico de una niña de 10 años diagnosticada con el síndrome de Rett, que muestra regresión en habilidades de comunicación y motricidad a partir de 18-36 meses. Se detallan criterios de diagnóstico desde las clasificaciones DSM-IV y CIE-10, así como las etapas del desarrollo del síndrome y sus implicaciones clínicas. El tratamiento es esencialmente paliativo, y un diagnóstico preciso es vital para mejorar la calidad de vida de los pacientes.


























































































