
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (17)
Destacado
Destacado (20)
Similar a Estabilidad química
Similar a Estabilidad química (20)
Último
Último (20)
Estabilidad química
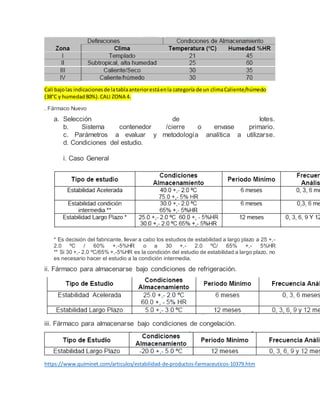
- 1. Cali bajolas indicacionesde latablaanteriorestáenla categoría de un climaCaliente/húmedo (38°C y humedad80%).CALI ZONA 4. . Fármaco Nuevo a. Selección de lotes. b. Sistema contenedor /cierre o envase primario. c. Parámetros a evaluar y metodología analítica a utilizarse. d. Condiciones del estudio. i. Caso General * Es decisión del fabricante, llevar a cabo los estudios de estabilidad a largo plazo a 25 +,- 2.0 ºC / 60% +,-5%HR o a 30 +,- 2.0 ºC/ 65% +,- 5%HR ** Si 30 +,- 2.0 ºC/65% +,-5%HR es la condición del estudio de estabilidad a largo plazo, no es necesario hacer el estudio a la condición intermedia. ii. Fármaco para almacenarse bajo condiciones de refrigeración. iii. Fármaco para almacenarse bajo condiciones de congelación. https://www.quiminet.com/articulos/estabilidad-de-productos-farmaceuticos-10379.htm
- 2. TALLER 1. Estabilidad: Capacidad de un ingrediente farmacéutico activo o producto farmacéuticoterminado, de mantener a través del tiempo sus propiedades originales dentro de las especificaciones establecidas, en relación a su calidad, seguridad y eficacia (por ejemplo identidad, concentración o potencia, pureza y apariencia física, etc.) (OMS). Estabilidad física: modificaciones en las características físicas. Estos cambios no llevan directamente a la degradación del fármaco, pero si implican la modificación de las características biofarmaceuticas del mismo en la formulación. Susceptibles: apariencia, contenido de humedad, velocidad de disolución, tiempo de disgregación, resistencia a la fractura y friabilidad (comprimidos). Se puede afectar la liberación del API y consecuentemente la biodisponibilidad de este (Ej: la biodisponiblidad de la aspirina oral esta entre 40-50%, y es mucho mayor si es intravenosa). 2. Cinética:Estudioque permite evaluarlavariaciónde laconcentracióndel principioactivo o Claimsrespectoal tiempo,donde se puede determinarlascausasy el mecanismo implicadoenel cambiodel producto. Estudioque permite evaluarlavariaciónde laspropiedadesfísicasyquímicasdel principio activoo Claimsrespectoal tiempo,donde se puededeterminarlascausasy el mecanismo implicadoenel cambiodel producto. 3. Factores de degradación asociados a: P. farmacéuticos: Humedad(H2O),luz,temperatura. Malascondicionesde almacenamiento, recipiente ymaterial usado. A. Incompatibilidad entre componentesde laformulaciónpre formulación Sustanciaácidacon sustanciaalcalina Sustanciacatiónicacon sustanciaaniónica Sustanciasoxidantesconsustanciasde carácter reductor Precipitación Formaciónde complejos B. Condicionesambientales Desarrollomicrobiano Humedad(alteraciónde formassolidas;acciónhidrolizante,oxidación, crecimientomicrobiano) Temperatura(aceleralosprocesosdegradativos,afectalascaracterísticasfísico- químicasdel medicamento) Oxígeno(Rx oxidaciónproduce radicaleslibres.Catalizadaspor:luz,metales pesadosycalor) Luz (Rx de fotolisis)
- 3. Cosméticos: Alteraciónde características organolépticas. Factoresextrínsecos (externosal producto):T,luz,humedad,oxigeno,y microorganismos. Factoresintrínsecos (dependende lafórmuladel producto):Incompatibilidad entre ingredientes,oxidacióne hidrolisis. Nota:La hidrolisisnotendrálugarencosméticossi estáenmedioaceitoso. La hidrolisisse puede vercomounavariacióndel PH. 4. Donde se puede ver afectaciónde la estabilidad: P. farmacéuticos: Operacionesunitarias: Granulaciónhúmeda:El usode solventesacuosospuede desencadenarRx de hidrolisis(Ej:degradacióndel A.acetilsalicílico). Lasoluciónesrealizarlasotras dos técnicassiguientesde tratamientoyenvasarse el medicamentoprotegidos con la humedad,esdecir,herméticos. Granulaciónseca:Se puedendarefectoshigroscópicosde losexcipientesoAPI. El golpe del polvopuede ocasionarel rompimientode interaccionesentre componentesnecesariosparamantenerunaformafarmacéuticaliquida, incidiendoenlaformaciónde precipitados. Compresióndirecta: Etapas de desarrollo: Antes,durante ydespués. Pre-formulación:
- 4. 5. Normas ICH: ICH ConferenciaInternacional sobre Armonizaciónde RequisitosTécnicos para el Registrode ProductosFarmacéuticosparaUso Humano que reúne alas autoridadesreguladorasylaindustriafarmacéuticade Europa,Japóny losEE.UU. Las basesdel nuevoMarco regulatorioaNivel mundialparalaindustriaFarmacéuticaestá sustentadoenlaserie de normasICH Q8, ICH Q9 e ICH Q10 : ICH Q8 : Calidaddel Diseñode ProductosFarmacéuticos. ICH Q9 : Gestiónde laCalidaddel Riesgo. ICH Q10: Sistemade CalidadFarmacéutico Importancia: La mejorade la saludmundial esel principal objetivoaconseguir,mediante la armonizaciónanivel internacionalde laproducciónde medicamentos,enordenasu seguridad,eficaciaydesarrollode altosestándaresde calidad. 6. Estudios de estabilidad:Evalúa las condicionesde almacenamiento. Condicionesde estréso extremas: Condicionesaceleradas o corto plazo: Estudiodiseñadoparaaumentarla velocidadde degradaciónquímicaocambiosenlaspropiedadesfísicasde una sustanciao un productofarmacéutico,empleando condicionesde almacenamientoextremas.Estosestudiostienencomoobjetodeterminarlos parámetroscinéticosde losprocesosde degradacióno predecirlavidaútil del productofarmacéuticoencondicionesnormalesde almacenamiento. Factores: Condicionesde Tyhumedadde almacenamiento Tiempode almacenamientoantesde lascorrespondientestomasde muestra Númerode lotesmuestreados Númerode replicadosde cadalote Influenciade laluz Detallesdel ensayo
- 5. Condicionesnaturales o larga duración(envejecimientonatural):Estudio diseñadoparala evaluaciónde lascaracterísticasde estabilidadfísica,química, biológicaymicrobiológicade unproductofarmacéuticoouna sustanciabajolas condicionesde almacenamientorecomendadas,que cubre todoel períodode vida útil ó el períodode re análisispropuesto. Estosestudiosse realizana“condiciones ambientales”.Objetivo:Establecerel tiempode caducidad de laformulación, considerandolasestimacionesrealizadasde los estudiosacelerados.
- 6. Monografía de la aspirina(ASA) 1. Por el grupo ésterque presente lamolécula essusceptible adegradaciónporhidrolisispor la presenciade agua. Además,tambiénpuede interferirenlaestabilidaddel fármacolos excipientesoaditivosque presente laformulacióncomo:los estearatos (de aluminio, magnesio,calcio) al mezclarse conlaASA,yaque aumentanlavelocidadde degradación de este.Porúltimo,laASA puede reaccionarcon aminasy gruposhidroxi,que producen lascorrespondientesamidas yesteres,respectivamente. Nota:El estearatode magnesiofacilitalatransferenciadel grupoaciloyaque acelera dichareacción. 2. Perfil cinéticode PH: Es un método graficopara determinarcomolacinéticade reacción, eneste caso degradación,de unamoléculase ve afectadaporlosdiferentesrangosde PH. En el graficose evalualogkvs PH y se tiene encuentael parámetrode la temperaturaen la cual se llevaacabo el experimento. Coneste perfilse puede determinarel PHdonde se dan lascatálisisacida,básica;además,el rangode PH-independientede K(donde nohay variaciónde k cuandovaria el PH enese rango) y finalmente,determinarel PHdonde se obtiene lavidamedia(𝑡1/2) yel tiempode caducidad(𝑡90). Importancia:Es esencial paradeterminarel PHde máximaestabilidad(puntomás bajode lacurva) y esome garantizara hacermisposterioresestudioscinéticos disminuyendoel errorporel parámetrode PH. 3. Hoja 4. Productosde degradación: Señales: FTIR (EspectrometríaInfrarrojaconTransformadade Fourier):ASA;OH
- 7. 5. Efectodel solvente acuososobre lacineticade degradación: Dependedel tipode estudio que vaya a realizaryla susceptibilidadque tengami farmacoendegradarse.Enel caso de un analitoque se degrade porhidrolisisyauto-oxidaciónynecesitoevaluarlacineticapor la Rx de oxidacióndeboaislarmi reactivode aguapara que nose de la otra reaccióny afecte mi estudio. FALTA TERMINARLA IDEA 6. Metodosde estabilización: ASA: Estrategiafisicoquimica: Usarel PH de máximaestabilidad,manipularla temperaturasadecuadasde bajadegradación;si lapresentacióndel mediamento esen soluciónusarunamezclaetanol/aguadonde hallamayor% del alcohol (disminuye lacte de velocidadde degradación). Est. Farmacotecnia:Nousar excipientesque presentenaltahigroscopicidadenla formulación,evitarel usode estearatos,disminuirel contactoconagua, hacer una cubiertao peliculade latabletaparareducirsu % de degradación, bloqueode los gruposhidroxi poralquilación oacilación.Ensuspensiones se usafluidosde methylpolysiloxane paravehiculizarel ASA. Escogerlaformade administración mas efectiva,eneste casolastabletas yaque endisolucionespuededarse hidrolisis. Colocarel medicamentoenempaquessellados, compresióndirectao granulacionseca. Control de pH: Utilizar el pH óptimo de estabilidad. Limitaciones: solubilidad, actividad terapéutica del p.a. y compatibilidad fisiológica Uso de disolventes: Disminuye la velocidad por incremento de la polaridad del medio. Ej : EtOH, Glicerina, Sorbitol, etc. Formación de complejos: Disminuye la velocidad por impedimento estérico o por incremento de polaridad Inclusión del p.a. en micelas Modificación de la estructura del p.a.: Adicionando determinados grupos a la estructura: efecto estérico o polar. (Asegurar que las propiedades farmacológicas permanecen constantes) 7. Las condiciones de los estudios de estabilidad varían dependiendo de las zonas de fabricación puesto que se sabe que hay zonas más húmedas y calurosas y/o con presiones diferentes, las cuales pueden afectar no tanto la producción sino al almacenamiento del producto. Esto se puede ver influenciado en su tiempo de degradación, ya que ciertas temperaturas o condiciones, aun siendo muy pequeñas pueden afectar tanto a los productos de degradación (aumentando o disminuyendo su concentración o incluso creando nuevos productos no deseados que pueden llegar a ser tóxicos). Por eso, las normas de armonización dan una directriz de cómo se deben hacer dichos ensayos de estabilidad pero no pueden dar precisión a como se comportaran los productos en todas las condiciones.
- 8. 8. Para el casi de Colombia,existeunanormativade estudiosde estabilidadesque se debenseguir para el procesode comercializaciónde productosfarmacéuticos,estedocumentose denomina “GUIA PARA EL DESARROLLOY PRESENTACIÓN DELOS ESTUDIOS DE ESTABILIDADDE MEDICAMENTOS CONVENCIONALES”(convencionalespuestoque existe unaguíapara medicamentosbiológicos)del Ministeriode ProtecciónSocial,el cual tiene lospasosque se deben de seguir,loscualesson: 2. ENSAYOS DE ESTABILIDAD PARA INGREDIENTES FARMACÉUTICOS ACTIVOS. ........................................................................................................ 12 2.1. Aspectos generales ............................................................................... 12 2.2. Ensayos bajo estrés .............................................................................. 12 2.3. Selección de los lotes ............................................................................ 13 2.4. Sistema de envase y cierre .................................................................... 14 2.5. Especificación ........................................................................................ 14 2.6. Frecuencia de análisis ........................................................................... 14 2.7. Condiciones de almacenamiento ........................................................... 15 2.7.1. Caso General .................................................................................. 16 2.7.2. Ingrediente Farmacéuticos Activos destinados a ser almacenados en un refrigerador ........................................................................................... 17 2.7.3. Ingredientes Farmacéuticos Activos destinados a ser almacenados en un congelador ...................................................................................... 18 2.7.4. Ingredientes Farmacéuticos Activos destinados a ser almacenados por debajo de -20ºC. ................................................................................. 18 2.8. Aseguramiento de la estabilidad (compromisos) ......................... 18 2.9. Evaluación ............................................................................................. 19 2.10. Leyendas y etiquetado ......................................................................... 21 2.11. Estudios de estabilidad sobre la marcha (ongoing stability studies) 21 3. ENSAYOS DE ESTABILIDAD EN LOS PRODUCTOS FARMACÉUTICOS TERMINADOS (PFT). ...................................................................................... 23 3.1. Aspectos generales ............................................................................... 23 3.2. Objetivos del estudio de estabilidad ...................................................... 23 3.3. Generalidades sobre el diseño .......................................................... 23 3.4. Diseño e interpretación de estudios de estabilidad ............................ 24 3.5. Metodología analítica indicadora de estabilidad .................................... 25 3.6. Selección de lotes ............................................................................... 25 3.7. Envase primario y cierre del producto.................................................... 26 3.8. Especificaciones del PTF. ...................................................................... 27 3.9. Frecuencia de los ensayos (muestreos) ............................................... 28 3.10. Condiciones de almacenamiento ......................................................... 28 3.10.1. Caso general ................................................................................. 30 3.10.2. PFTs envasados en recipientes impermeables ............................. 32 3.10.3 PFTs envasados en recipientes semipermeables .......................... 32 3.10. 4. PFTS destinados a ser almacenados en un refrigerador ............. 35 3.10.5. PFTS destinados a ser almacenados en un congelador ............... 35 3.10.6. PFTS destinados a ser almacenados por debajo de – 20°C ......... 36 3.11. Aseguramiento de la estabilidad (compromisos) ....................... 36 3.12. Evaluación ........................................................................................... 37
- 9. 3.13. Leyendas y etiquetado ......................................................................... 39 3.14. Estabilidad en uso ............................................................................... 40 3.15. Variaciones .......................................................................................... 41 3.16. Estudios de estabilidad sobre la marcha (ongoing stability studies) .... 41 4. RECOMENDACIONES PARA EL DISEÑO DE ESTUDIOS DE ESTABILIDAD ................................................................................................. 43 4.1. Aspectos generales ............................................................................... 43 4.1.1 Fluctuaciones extremas de temperatura .......................................... 44 4.1.2. Temperaturas de almacenamiento ............................................... 44 4.1.3 Calidad Microbiológica .................................................................. 44 4.1.4 Productos de descomposición o degradación. .............................. 45 4.1.5. Consideraciones sobre el diseño comúnmente aceptadas de acuerdo con los ensayos a efectuar de acuerdo a la forma farmacéutica en estudio. ...................................................................................................... 46 4.1.6. Consideraciones de tipo estadístico ............................................ 51 4.2. Diseños de estudios de estabilidad de largo plazo ................................ 52 4.2.1. Consideraciones para el Diseño experimental ............................. 52 4.2.2. Consideraciones para el muestreo del lote .................................. 53 4.2.3. Consideraciones sobre el número de lotes a utilizar .................... 53 4.2.4 Consideraciones a aplicar sobre el muestreo de acuerdo con el envase primario y el cierre del producto.................................................... 54 4.2.5. Consideraciones sobre el número de muestras a utilizar ............. 56 4.2.6 Consideraciones sobre el tiempo de muestreo ............................. 58 4.2.7. Análisis de los datos e interpretación de los estudios de estabilidad de largo plazo .......................................................................... 59 4.3. Estudios de estabilidad bajo otras condiciones ................................... 65 4.3.1 Tipos de estudios según el objetivo propuesto ............................ 65 4.4. Establecimiento de las condiciones de un estudio de estabilidad ....... 67 4.4.1 Efecto de la temperatura ............................................................... 67 4.4.2 Efecto de la Humedad Ambiental .................................................. 68 4.4.3 Efecto de la combinación de Humedad y Temperatura Ambientales .................................................................................................................. 68 4.4.4 Efecto de la luz .............................................................................. 68 4.5. Recomendaciones para los estudios de Estabilidad Acelerada ó Envejecimiento acelerado ............................................................................. 68 4.6. Definición del producto bajo estudio .................................................... 71 4.6.1 Cantidad y naturaleza de los lotes evaluados .............................. 71 4.6.2 Envase primario o inmediato ........................................................ 71 4.7. Características a evaluar ..................................................................... 72 4.7.1. Características físicas y aspectos microbiológicos del PFT ........ 72 4.7.2. Características químicas del producto terminado......................... 73 4.7.3 Características del empaque a ser consideradas......................... 73 4.8. Consideraciones sobre los métodos de evaluación ............................. 73 4.9. Presentación de resultados .................................................................. 73 4.10. Discusión e interpretación de los resultados y conclusiones ............. 74 5. PRECISIONES SOBRE EL DISEÑO DE ESTUDIOS DE ESTABILIDAD . 75 5.1. Estudios de estabilidad acelerada ......................................................... 75 5.1.1. Número de lotes a evaluar ........................................................... 75
- 10. 5.1.2. Tiempo .......................................................................................... 75 5.1.3. Condiciones del estudio ............................................................... 75 5.1.4. Tipos de Métodos o Diseños Experimentales aceptados ............. 76 5.2. Estudios de estabilidad a largo plazo................................................. 82 5.2.1. Número de lotes a evaluar ........................................................... 82 5.2.2. Tiempo ......................................................................................... 82 5.2.3. Condiciones del estudio ............................................................... 82 5.2.4. Tiempos de muestreo ................................................................... 82 5.2.5. Manejo de los datos obtenidos ..................................................... 83 5.2.6. Tipos de Métodos o Diseños Experimentales aceptados ............. 83 5.2.7. Presentación y evaluación de los resultados ............................... 84 6. PRESENTACIÓN DEL INFORME DEL ESTUDIO DE ESTABILIDAD ....... 84 7. Las condiciones de los estudios de estabilidad varían dependiendo de las zonas de fabricación puesto que se sabe que hay zonas más húmedas y calurosas y/o con presiones diferentes, las cuales pueden afectar no tanto la producción sino al almacenamiento del producto. Esto se puede ver influenciado en su tiempo de degradación, ya que ciertas temperaturas o condiciones, aun siendo muy pequeñas pueden afectar tanto a los productos de degradación (aumentando o disminuyendo su concentración o incluso creando nuevos productos no deseados que pueden llegar a ser tóxicos). Por eso, las normas de armonización dan una directriz de cómo se deben hacer dichos ensayos de estabilidad pero no pueden dar precisión a como se comportaran los productos en todas las condiciones. DIAGRAMAS DE ENERGÍA POTENCIAL
- 11. La reacciónes exotérmicay el ∆G en este casoes negativo. ∆G=(+) Rx endotérmica ∆G=(+) Rx exotérmica El primerestadode transiciónesel pasodeterminante de lavelocidadde reacción,porsermás energético,esdecir,unaaltaEa para que se dé. A diferenciade losestradosde transiciónque no son aislables,losintermediariossípuedenaislarse,tienentiemposde vidamediafinitosyson más establesque losestadosde transición,peromenosque losreactivosoproductos.Losdiagramas de energíapotencial sonútilesenladiscusióncualitativade losmecanismosde reacción. Diagrama de energía La primera etapa de propagación es la que limita la velocidad del proceso, tiene la mayor energía de activación. El el diagrama se representan reactivos, productos, intermedios y estados de transición para la halogenación radicalaria del metano.
- 12. En el primer estado de transición se forma el enlace H-Cl y se rompe el C-H. En el segundo estado de transición se forma el enlace C-Cl y rompe el Cl-Cl.