Descargar para leer sin conexión



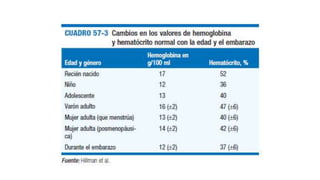
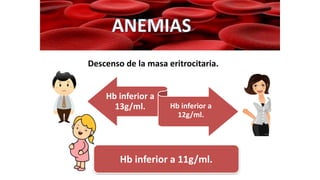

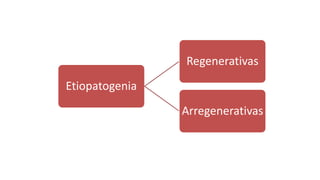

















































1. El documento presenta información sobre diferentes tipos de anemias, incluyendo sus causas, síntomas, exámenes de laboratorio y tratamientos. 2. Se describen anemias como la ferropénica, megaloblástica, hemolítica y otras, proporcionando detalles sobre su etiología, manifestaciones clínicas y abordaje diagnóstico y terapéutico. 3. El resumen brinda una visión general de los principales tipos de anemias discutidos en el documento de manera concisa.















































![ANEMIA GENERALIDADES.pptx [Reparado] (1).pptx](https://cdn.slidesharecdn.com/ss_thumbnails/anemiageneralidades-250927041105-ef7a9965-thumbnail.jpg?width=640&height=640&fit=bounds)





























