Descargado 21 veces




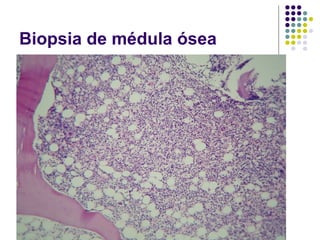











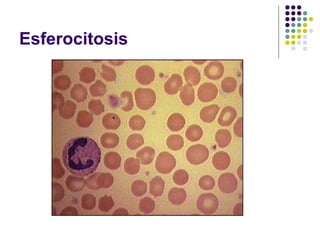










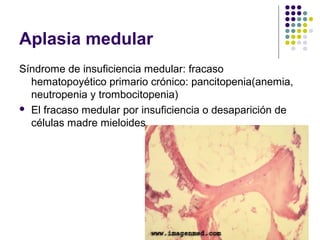














El documento describe diferentes tipos de anemias, incluyendo anemias hemolíticas, esferocitosis hereditaria, deficiencia de glucosa-6-fosfato deshidrogenasa, enfermedad de células falciformes, aplasia medular, y anemias megaloblásticas como la anemia perniciosa y deficiencia de ácido fólico. Explica las causas, morfología, curso clínico y características diagnósticas de cada tipo de anemia.


































































