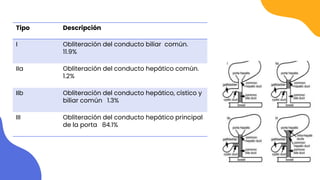
Este documento presenta información sobre colestasis neonatal. Define la colestasis como una disminución en la formación o flujo de bilis que genera retención de sustancias biliares en el hígado. Detalla la epidemiología, abordaje diagnóstico incluyendo exámenes, y posibles causas como atresia de vías biliares, quiste de colédoco, síndrome de Alagille, infecciones y errores metabólicos.