Descargado 89 veces

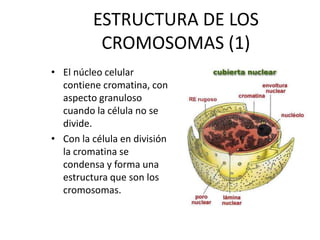

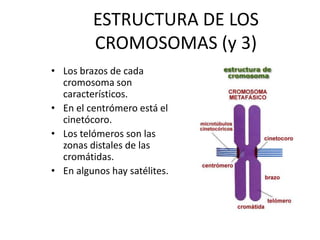

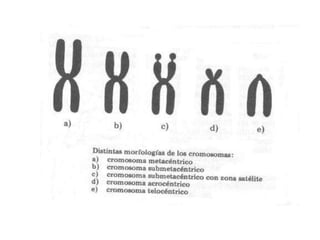
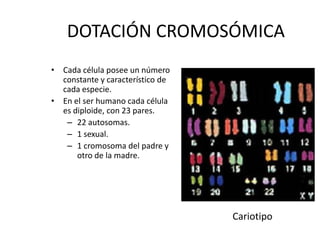


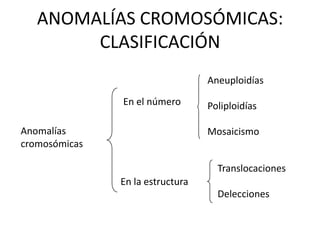



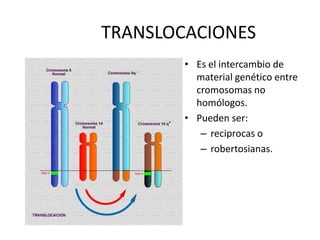
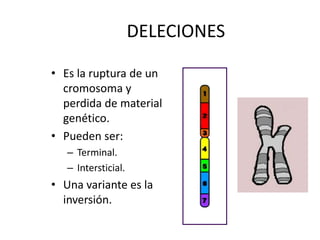



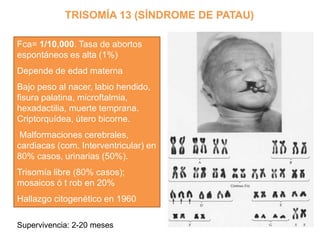
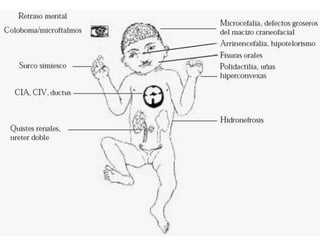

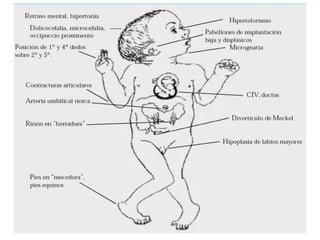

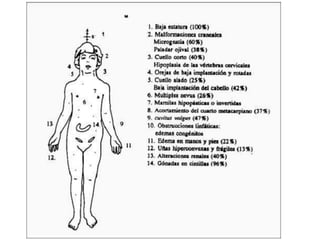

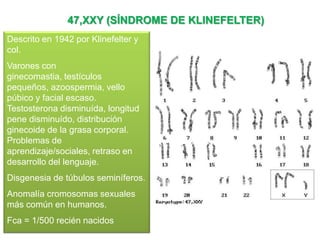
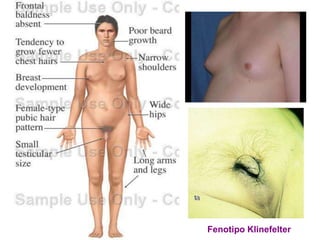
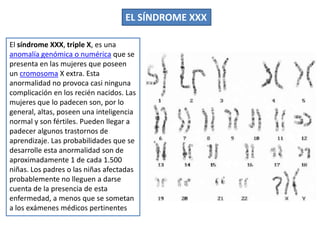


El documento describe la estructura y tipos de cromosomas, así como diferentes enfermedades genéticas asociadas con anomalías cromosómicas. Explica que los cromosomas están formados por ADN y proteínas y contienen los genes. Las células humanas normalmente contienen 23 pares de cromosomas. Las enfermedades genéticas pueden deberse a cambios en el número o estructura de los cromosomas, como en los síndromes de Down, Patau, Edwards, Turner y Klinefelter.























































































