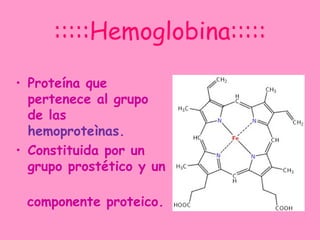
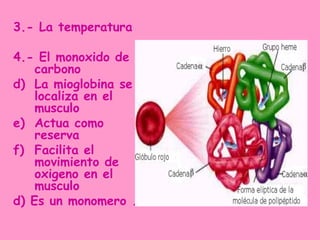
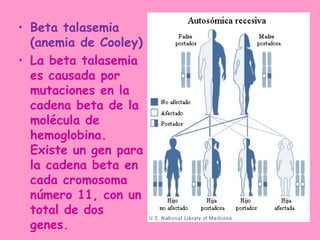
La hemoglobina es una proteína que transporta oxígeno en la sangre. Está compuesta por un grupo hemo y una parte proteica. El grupo hemo contiene hierro y se forma a través de la unión de varios compuestos, incluyendo succinil CoA. La hemoglobina tiene cuatro cadenas polipeptídicas y transporta oxígeno de manera cooperativa. Existen diferentes tipos de hemoglobina en diferentes etapas de la vida, como la hemoglobina fetal y la hemoglobina A del adulto.