Descargado 45 veces












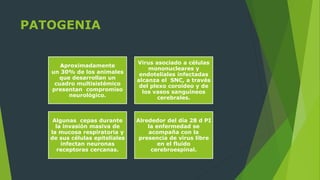





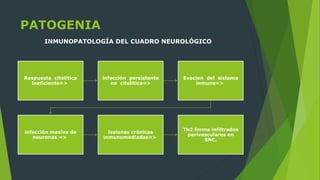








El documento describe la patogenia del virus del distemper canino. El virus ingresa a través de las mucosas y se replica rápidamente en los linfocitos, lo que causa una supresión del sistema inmune. Esto permite la diseminación sistémica del virus y el establecimiento de cuadros multisistémicos. El virus también puede infectar el SNC, causando encefalitis mediante mecanismos tanto citopáticos como inmunopatológicos que conducen a desmielinización y daño neuronal.
































































![Enfermedades Desmielinizantes[1]](https://cdn.slidesharecdn.com/ss_thumbnails/enfermedadesdesmielinizantes1-1214281635926917-8-thumbnail.jpg?width=640&height=640&fit=bounds)









![Vacunas de dna[1]......](https://cdn.slidesharecdn.com/ss_thumbnails/vacunasdedna1-141216144414-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)


























