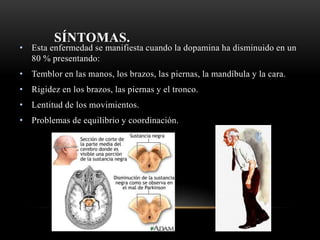
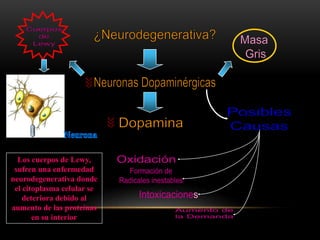
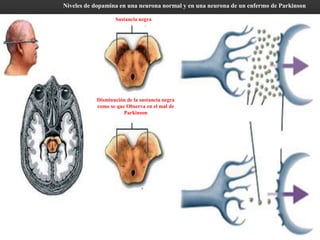
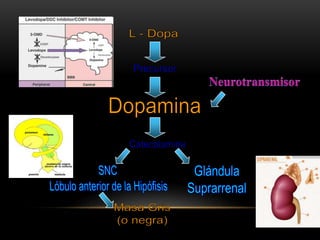
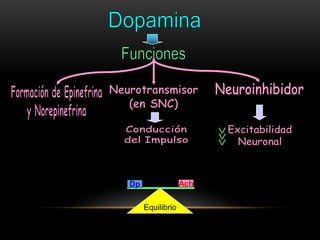
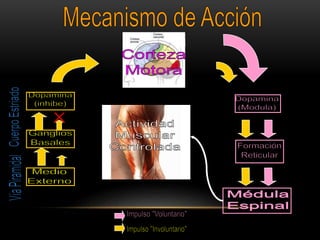
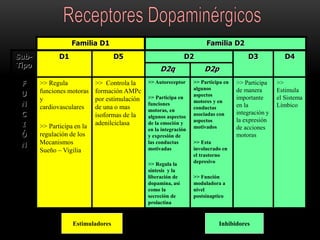
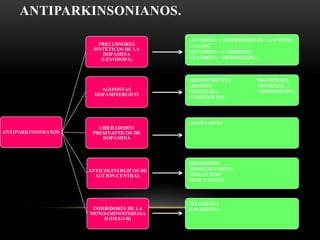
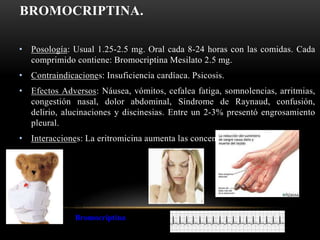
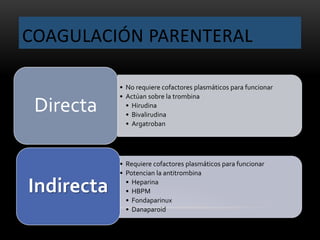
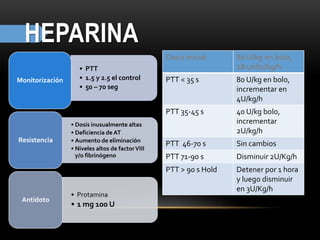
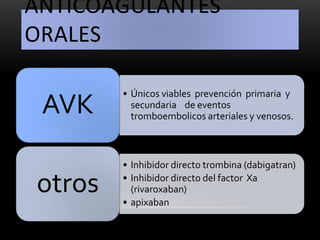
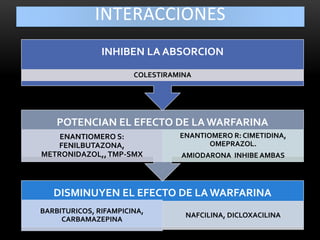
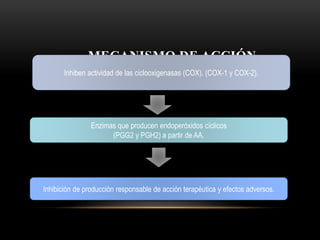
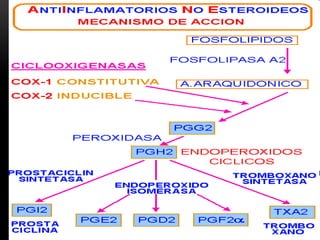
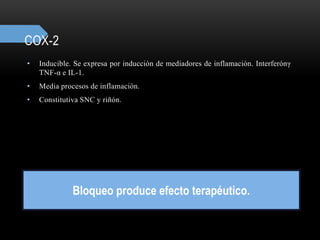
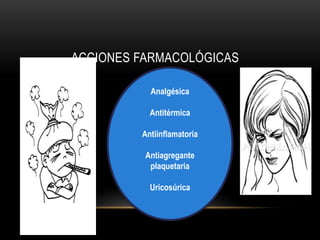
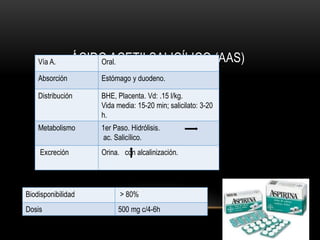
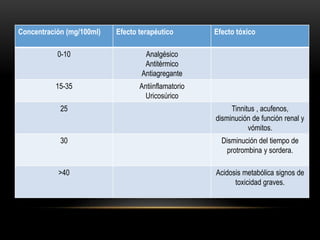
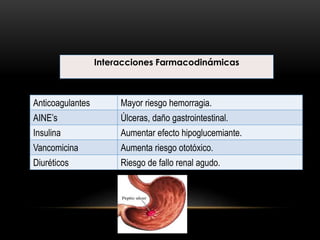
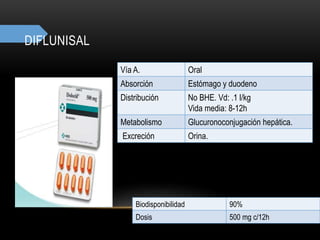
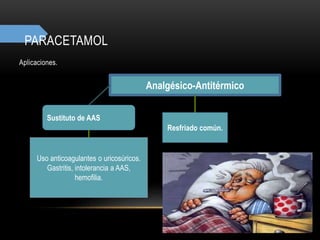
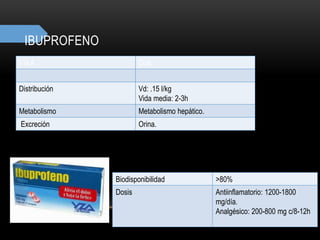
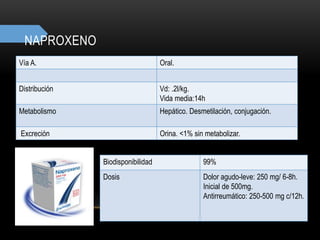
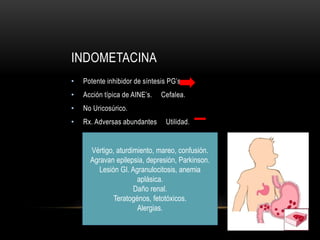
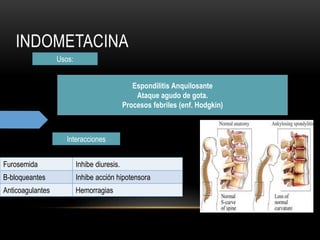
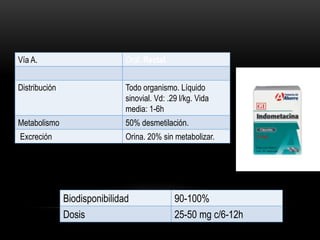
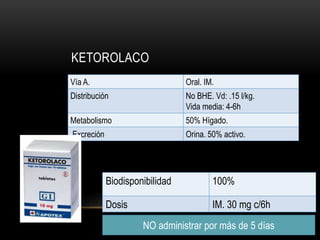
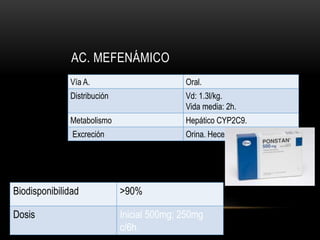
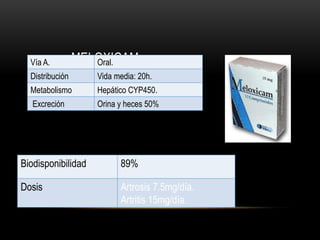
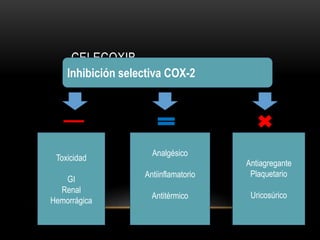
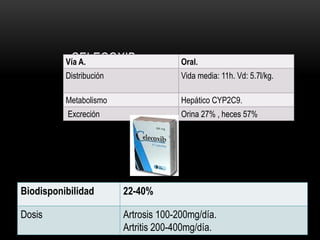
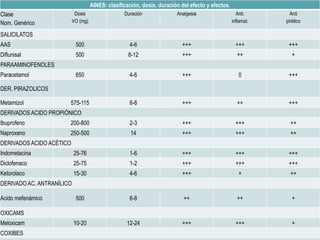
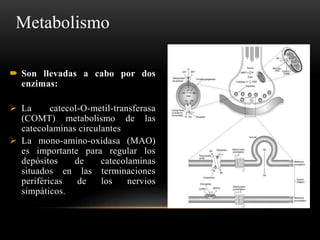
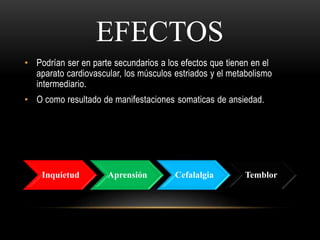
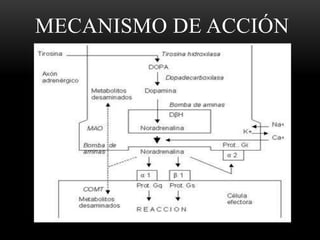
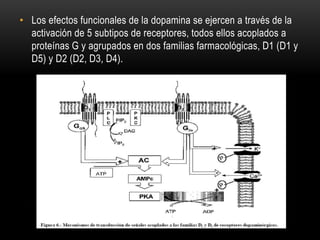
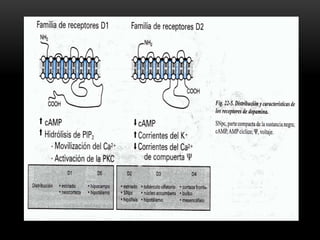
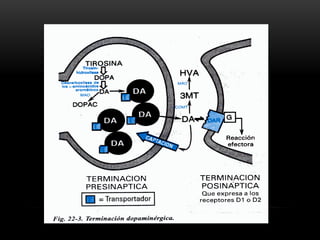
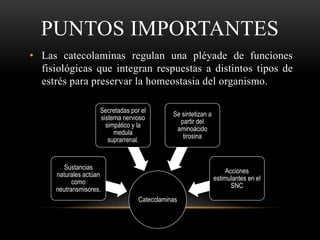
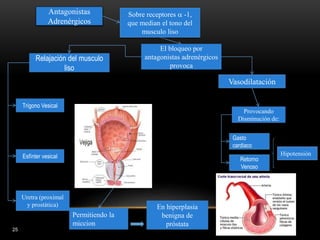
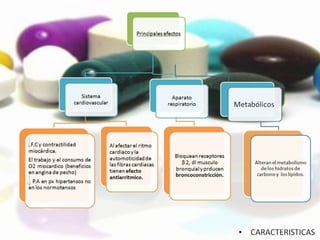
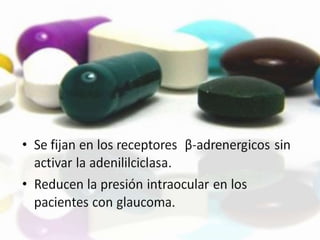
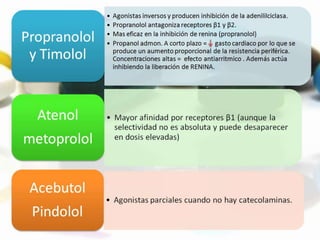
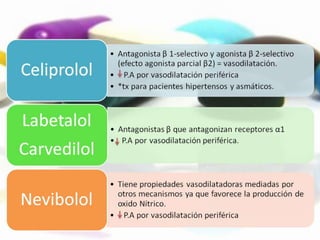
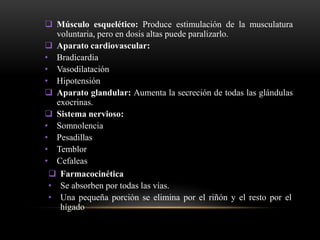
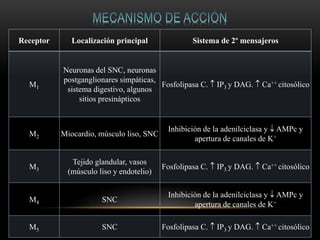
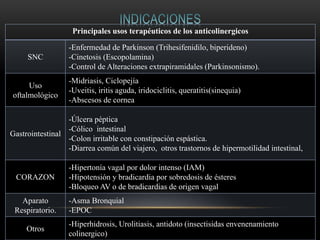
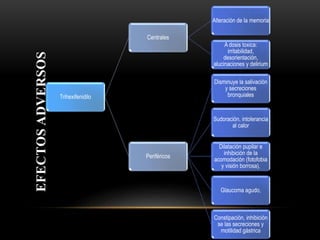
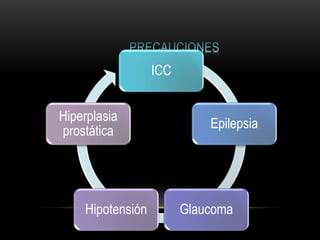
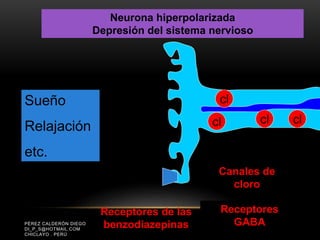
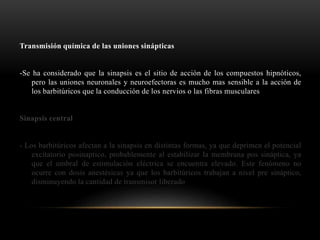
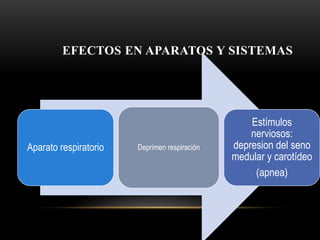
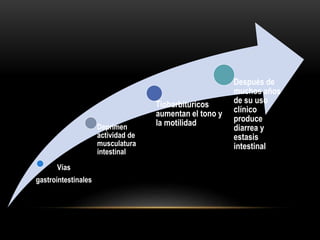
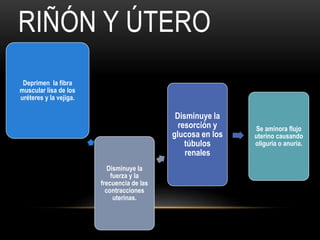
Las catecolaminas son sustancias naturales que actúan como neurotransmisores. Se sintetizan a partir del aminoácido tirosina y son secretadas por el sistema nervioso simpático y la médula suprarrenal. Incluyen la dopamina, noradrenalina y adrenalina, y regulan funciones fisiológicas para preservar la homeostasis. Se unen a receptores en la neurona postsináptica para ejercer sus efectos estimulantes en el SNC, aunque no cruzan fácilmente la barrera hematoencefá




























































































































































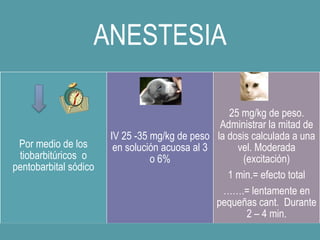






















































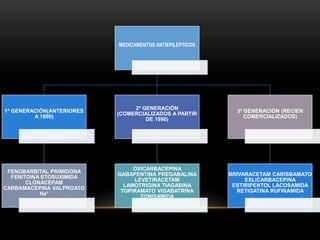








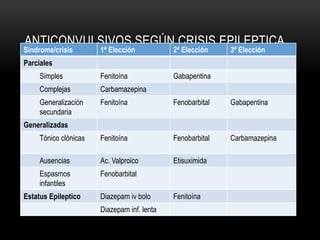
![CONVULSIONES PARCIALES
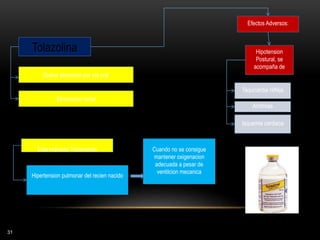
Simples
Sin pérdida de conciencia
[20↔60]segundos
Clínica vs área cortex activada
Complejas
↓ conciencia
[30segundos ↔ 2minutos]
Movimentos sin objetivo
Generalizadas
Pérdida de conciencia
[1↔2]minutos
Contracción muscular mantenida
(fase tónica), seguida de períodos
de contracción-relajación (fase
clónica)
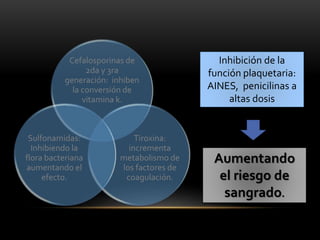
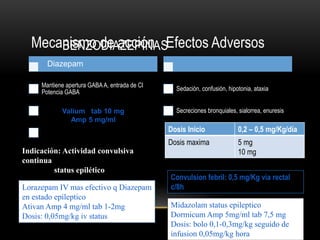
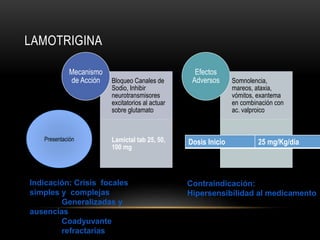
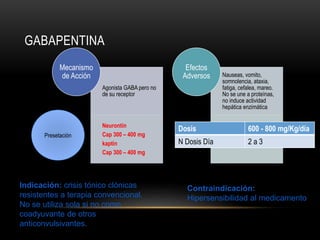
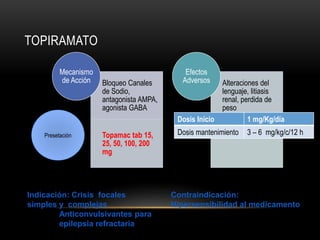
CBZ
FNT
VAL
CBZ
FNB
FNT
PRIM
VAL
GAB
LAM
LEV
RUF
TIA
TOP
ZON](https://image.slidesharecdn.com/completo-150225131657-conversion-gate01/85/FARMACOLOGIA-4TO-SEMESTRE-COMPLETO-280-320.jpg)
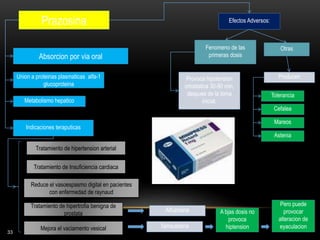
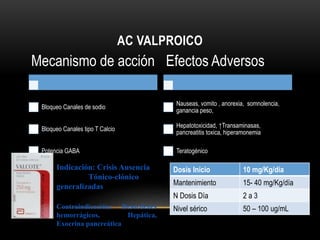
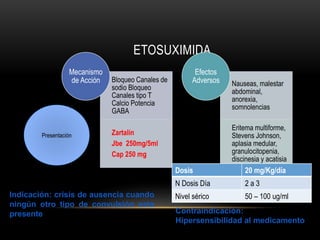
![CONVULSIONES GENERALIZADAS
Crisis de ausencia
Fallo cognitivo, asociado con
fijación de la mirada y bloqueo de
actividades motoras
[<30 segundos]
Mioclonias
Breve contraccíón muscular con
afectación variable
A veces < 1 segundo
Convulsiones
Tónico-clónicas
Convulsiones tónico-clonicas que
no van precedidas por una
convulsión parcial
EXS
VAL
CNZ
VAL
CNZ
LAM
CBZ
FNB
FNT
PRIM
VAL
LEV
LAM
LEV
TOP](https://image.slidesharecdn.com/completo-150225131657-conversion-gate01/85/FARMACOLOGIA-4TO-SEMESTRE-COMPLETO-281-320.jpg)
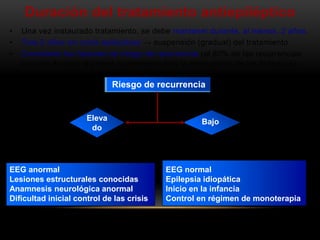
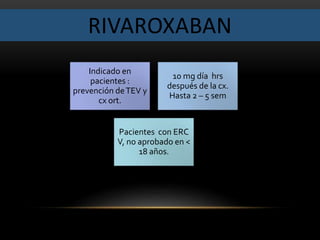
![ANTIEPILÉPTICOS Y EMBARAZO: CONSEJOS
• Preferible consejo antes de la concepción: consejos
sobre riesgos genéticos, interacciones con
contraceptivos orales, diagnóstico prenatal (médico
de familia, neurólogo experto en epilepsia,
ginecólogo, genetista y pediatra).
• Posibilidad de retirar la medicación antiepiléptica con
seis meses de antelación a la planificación de la
concepción (si la mujer ha estado entre 2 y 5 años
sin crisis convulsivas, y con limitados factores de
riesgo).
• Si ha de continuar con la medicación:
• Intentar la monoterapia (evitar asociaciones de varios
medicamentos antiepilépticos).
• Evitar formulaciones de uso único diario (evitar CMÁX
elevadas asociadas a importante fluctuación diaria de
las concentraciones, sobre todo si se usa Valproato).
• Administrar ácido fólico a dosis elevadas (4g/día)
[sobre todo con Carbamacepina o Valproato].
• Si el embarazo no se ha planificado, continuar con el
tratamiento establecido. Si aparecen crisis, hay que
considerar:
• Incumplimiento del tratamiento por la paciente.
• Modificación de las concentraciones en suero.
Fetos: semana 14ª
(arriba); 28ª (abajo)](https://image.slidesharecdn.com/completo-150225131657-conversion-gate01/85/FARMACOLOGIA-4TO-SEMESTRE-COMPLETO-283-320.jpg)
![TRATAMIENTO ANTIEPILÉPTICO Y EMBARAZO
• Efectos teratógenos asociados a
antiepilépticos:
• Defectos cardíacos congénitos.
• Cierre incompleto del canal medular
(defectos de tubo neural).
• Labio leporino.
• Paladar leporino.
• Riesgo de malformaciones:
• [2%↔4%] población general versus
[4%↔8%] mujeres epilépticas bajo
tratamiento.
• Los antiepilépticos:
• ↓ eficacia de los anticonceptivos orales
(por inducción de la actividad enzimática
hepática responsible de su metabolización).
• ↓ la actividad anticoagulante de la
vitamina K → ↑ riesgo de hemorragia y
coagulopatía intracerebral.](https://image.slidesharecdn.com/completo-150225131657-conversion-gate01/85/FARMACOLOGIA-4TO-SEMESTRE-COMPLETO-284-320.jpg)
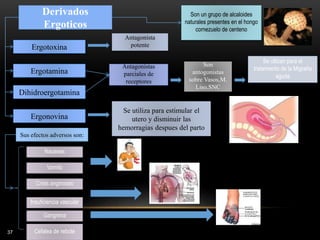
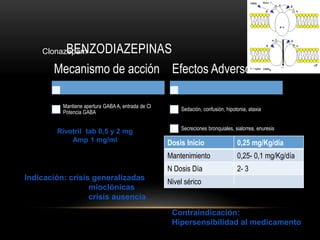
![CONVULSIONES FEBRILES
[2%↔4%]
[25%↔33%]
[2%↔3%]
1ª crisis convulsiva
2ª crisis convulsiva
Niños epilépticos
Anamnesis neurológica
Historia familiar epilépticca
Duración crisis > 15 minutos
2ª crisis el mismo día
Niños con riesgo elevado: Diacepam rectal en caso de fiebre ≥ 38º](https://image.slidesharecdn.com/completo-150225131657-conversion-gate01/85/FARMACOLOGIA-4TO-SEMESTRE-COMPLETO-285-320.jpg)